
Journal of Entrepreneurial Health Sciences and Innovation (JEHSI)™
From the Anticipated Role of Leucine Aminopeptidase and
SARS-CoV-2 Adaptive Evolution by Clonal Interference to the
Clinical Significance of Emerging Variants and Vaccination
Strategies against COVID-19
Dr Gabriel Pulido-Cejudo1,2*, Peter Humphries2, Dr Bratati Kar1,2, Dr Khadija El Abdaimi1,2 and Dr Abraham Pulido-Cejudo3
1International Centre for Advancement of Health Regional Innovation and Science, ICAHRIS Research
*corresponding author, gabriel.pulido-cejudo@icahris.org
2Canadian Federation of Breast Diseases, CFBD
3Hospital General de México
Abstract
In one of our early reports addressing the emergence and possible multifactorial origins of the S (Serine) and L (Leucine) variant types of SARS-CoV-2, we estimated relatively high mutation rates for SARS-CoV-2 types being equivalent to ≥ 6.0 x 10-6 nucleotide substitutions per site daily or 0.2 mutations per genome per day. Taking into account the former SARS-CoV-2 estimated mutation rate, together with the incidence of close to 200 million individuals infected globally and no effective antiviral treatments available, the recent unprecedented emergence of promising vaccination strategies to hamper infectivity, transmission and fatalities due to COVID-19 could be potentially challenged by the continuous rapid emergence and spreading of more aggressive SARS-CoV-2 variant types due to the high levels of global infections and transmission. This equally makes possible the co-infectivity with more than one viral variant type present in infected individuals as well as the presence of more than one mutation of relevance within the same variant. In this article, we describe current phylogenetic analysis of SARS-CoV-2, salient characteristics of variants of concern and, most importantly, the possible genomic adaptive evolution of SARS-CoV-2 through processes such as clonal interference and molecular convergent evolution of selective beneficial mutations. Based upon the proven ability of a SARS-CoV-2 polybasic Furin mutant lacking the Furin (S-Pro-Arg-Arg-Ala-R/PRRA) cleavage site motif (ΔPRRA) to elicit attenuated infections both in human cells and in animal models, we propose the role of selective aminopeptidases belonging to the M1 family, such as Leucine Aminopeptidase (LAPase), as immediate viral entry accessory receptor(s) (IVEARs) independent of the ACE2/Neuropilin-1-dependent pathway. We equally emphasise the urgent need to consistently perform field-validated population surveillance and genotyping concerning current and emerging variants, particularly searching for the presence of viral variant co-infectivity in SARS-CoV-2 infected individuals and its potential impact in recent vaccination campaigns. The necessity of raising awareness amongst vaccinated populations of their potential to become infected and transmit SARS-CoV-2 variants to vaccinated and unvaccinated populations is also underlined. Based upon potential concurrent intracellular viral pathways of SARS-CoV-2, the need for the use of potentially beneficial adjuvant biotherapeutics compounds, in particular of competitive Leucine/Arginine Aminopeptidase (LAPase) inhibitors and gut microbiota nNE biogenic clones, to increase the effectiveness of ongoing vaccination strategies to halt SARS-CoV-2 transmission and infectivity is also discussed. A brief review of current vaccine types, including their safety and efficacy against variants of concern, is also summarised.
Keywords: COVID-19, SARS-CoV-2 variants, mutation rates, SARS-CoV-2 variant co-infectivity, SARS-CoV-2 variant super-spreaders, asymptomatic super-spreader, intraspecies transmission, interspecies transmission, zoonotic transmission, viral load, nNE adjuvant biogenic clones, SARS-CoV RaTG13, hibernaculum, Angiotensin Converting Enzyme 2 (ACE2), immediate viral entry accessory receptors (IVEARs), Neuropilin-1, Receptor Binding Motif (RBM), S1-S2 subunits, Furin (S-Pro-Arg-Arg-Ala-R) polybasic cleavage site, mutation rates, multifactorial model, gene editing, lysosomotropic agents, SARS-CoV-2/(ACE2)-pH-dependent endocytosis, Post-Acute COVID-19 Syndrome, hyperinflammation
Article
Introduction
Most likely, December 8, 2019, signalled the beginning of one of the most substantial human health challenges in decades, with the emergence of probably one of the first clinical reports of patients suffering from pneumonia of unknown origin, now undoubtedly identified as one of the first documented cases of SARS-CoV-2/COVID-19 infections[1-6].
In one of our early reports addressing the emergence and possible multifactorial origins of the S (Serine) and L (Leucine) variant types of SARS-CoV-2[4], we estimated relatively high mutation rates for SARS-CoV-2 types being equivalent to ≥ 6.0 x 10-6 nucleotide substitutions per site daily or 0.2 mutations per genome per day[4].
On the basis of this estimate, as well as on detailed functional and structural analysis of SARS-CoV-2, together with the lack of an assertive characterisation of an intermediate animal viral reservoir, we advanced the concept of a direct zoonotic transmission from bats to humans and subsequent asymptomatic human-to-human super-spreader-mediated transmission and adaptive evolution[1-4].
Our dynamic multifactorial model on the origins of SARS-CoV-2 and its estimated relatively high mutation rate also provided an explanation for the initial rapid dominance of the L (Leucine) viral type concomitant with the gradual displacement of the ancestral SARS-CoV-2 S (Serine) viral type[1]. In addition, it also underscored the basis for the expected rapid and global emergence of numerous other variants, asymmetrically affecting several countries during recent months, carrying, among others, the SARS-CoV-2 spike E484K mutation[1-10].
The elucidation of the exact events preceding the emergence of SARS-CoV-2 have been hampered by the absence of ancestral strains isolated either from animal reservoirs or early human samples containing signature single nucleotide variations (SNVs) that encompass critical sequences required for SARS-CoV-2 optimal Angiotensin Converting Enzyme 2 (ACE2)-pH-dependent binding, endocytosis and proteolytic processing through polybasic Furin (S-Pro-Arg-Arg-Ala-R) cleavage site[1,4,11-13].
Nonetheless, elegant and rather encompassing experiments related to the use of a reverse genetic system to generate a SARS-CoV-2 strain devoid of (S-Pro-Arg-Arg-Ala-R; ΔPRRA) capable of eliciting an in vivo attenuated infection sustain our previous proposal of a plausible direct infectivity of bat SARS-related-CoV (SARSr-CoV) RaTG13 to humans leading to a primordial attenuated infection[4,14].
In this article, we further propose that human-to-human super-spreader-mediated transmission eliciting an attenuated human infection could have lead to the emergence of the ancestral SARS-CoV-2 S (Serine) variant type, giving rise to the more prevalent infectious L (Leucine) variant through synonymous and non-synonymous mutations and selection through clonal interference similar to that documented in other RNA viruses[15].
In the course of further developing this possible evolutionary pathway of SARS-CoV-2, we performed an in-depth analysis of publicly available known strains, summarising key phylogenetic viral genome sequence analysis reported to date in accordance with both Nextstrain and PANGO database lineages and emerging variants.
A particular effort was made to reconcile information concerning the reporting of initially described strains such as MT291826, first characterised on December 30, 2019, with current standardisation and reporting of emerging variants, looking for other possible ancestral viral sequences of interest that could have lead to the emergence of SARS-CoV-2 S (Serine) and L (Leucine) variant types.
Finally, taking into account the ability of the ΔPRRA SARS-CoV-2 strain, devoid of the Furin polybasic cleavage site (S-Pro-Arg-Arg-Ala-R), to elicit an in vivo attenuated infection not only supports our view of a potential direct infection from a bat Sarbecovirus such as bat coronavirus RaTG13 or, possibly, bat coronavirus Rm YN02, it equally supports the possible involvement of concurrent alternative intracellular viral pathways of SARS-CoV-2, enabling immediate viral entry and intracellular processing.
Based upon reported sequences at the junction of S1/S2 subunits of SARS-CoV-2 receptor binding domain[16] and our previous description and characterisation of accessory receptors for HIV immediate viral entry, we propose that Leucine Aminopeptidase could play a role in SARS-Cov-2 infectivity during immediate early events of receptor binding and viral-receptor complex processing[17-19].
First Set of Ancestral and Recent SARS-CoV-2 Global Variants and their Implications to Global Human Health
Although the international scientific community responded with a concerted and extraordinarily fast pace at the reporting of the onset of a group of seven patients suffering from acute pneumonia of unknown aetiology in Wuhan, the capital city of the Hubei province in central China[1-4,6,20], with few exceptions, the combined public health efforts articulated between global health organisations, public health officials and decision makers was insufficient to hinder the rapid emergence and spreading of more challenging SARS-CoV-2 variants. From the scientific and clinical research standpoint, the outpouring of valuable information calls for the continuous consolidation and standardisation of genomic, functional, structural and regional pathophysiological mapping and characterisation of emerging SARS-CoV-2 variant outbreaks.
Signature variation clusters elucidated from single nucleotide variations (SNVs) performed in tandem using approximately 2, 6 and 40 x 103 complete SARS-CoV-2 genome sequence analysis in protein coding and 5' untranslated regions have unveiled six distinct strain types (Type I-VI [VIa,b])[21]. Interestingly, Type II contains SNVs C8782T (Serine) and T28144C (Leucine), which were also used to define the S (Serine) and L (Leucine) types of SARS-CoV-2, respectively, equally encompassing their corresponding non-synonymous point mutations, C28144 (Serine)/T28144 (Leucine)[22].
Excitingly, the SNVs C8782T (Serine) and C28144C (Serine) first co-appeared in strain MT291826 of SARS-CoV-2 which, reported as early as December 30, 2019, represents one of the earliest SARS-CoV-2 variant strains found in China belonging to Clade L84S[23]. Apart from being detected in, perhaps, one of the first SARS-CoV-2 variants (MT291826) in addition to that originally isolated on December 26, 2019, and described under the name of WH-Human 1 coronavirus[24], the two aforementioned Single Nucleotide Variations (SNVs) possess a coefficient of allelic association of R2 = 0.987[21].
Based upon this strong allelic association, it is plausible that these early SNVs were co-transmitted in the course of initial and subsequent infections, most likely with concurrent or sequential incidences leading to in vivo asymmetrical mutation patterns of ancestral forms of SARS-CoV-2 that were most likely close to the S (Serine) ancestral Type[4,21,22]. Comparative analysis of SNVs within Type II variants, including the S (Serine) Type of SARS-CoV-2, against those found in various animal coronavirus strongly suggests that the S (Serine) Type is not only closer to bat SARSr-CoV RaTG13 but that it is, most likely, closer to the original strain of SARS-CoV-2 infecting humans[4,21,22].
During the early phases of the ongoing pandemic outbreak, from December 2019 to March 2020, it was fairly well established that SARS-CoV-2 S (Serine) and L (Leucine) Types accounted for 30% and 70% of the total number of SARS-CoV-2 global infections, respectively[21,22]. Around January 24, 2020, variants carrying, amongst others, the D614G mutation clustered within the Type VI group became the dominant group in the world[21].
Although we will proceed to describe some of the most salient characteristics of the six strain Types of SARS-CoV-2 based upon signature variation cluster analysis, a brief description of the most recent variants of interest and concern of SARS-CoV-2 is summarised in Table 1.
| First Set of SARS-CoV-2 Global Variants after Detection and Characterisation of the S (28144:ORF8) and L (8782:orf1ab) Types from the MT291826 Strain | ||||
| Common Current Variant Name / Representative Mutations | Region / Countries of Emergence | First Report | Countries Affected | Countries / Regions of Predominance |
| D614G | China, Europe, early predominant clade | Molecular dating analysis estimated the emergence of this clade to have taken place around mid-to-late January (January 10-25), 2020. | Global with current changes in geographical frequencies. | Global, previously predominant in Europe and North America. Apparent higher frequencies and viral load in younger COVID-19 Patients in the UK[25]. |
| 20A.EU1 / C22227T, C28932T, G29645T A222V | Spain, Netherlands and other Western European countries such as the United Kingdom, Switzerland, Ireland, Belgium and Norway were part of first infectious cluster in the EU[26]. | Nascent sequences seem to have been detected around June 20, 2020, when 7 Spanish sequences and 1 Dutch sequences were analysed. | Spain, Netherlands, UK (England, Northern Ireland, Scotland, Wales), Switzerland, Ireland, Norway, Belgium, France, Sweden, Hong Kong, Germany, Latvia, Italy and New Zealand | Primarily Western Europe, Spain, Wales, Scotland, Ireland, Switzerland and England, predominance period June-October, 2020. |
| 20A.EU2 / C4343T, G5629T, G22992A S477N | France, United Kingdom, Netherlands, Switzerland, Belgium, Spain and Norway | Sequences first reported primarily in France at the beginning of July, 2020. | France, United Kingdom, Netherlands, Switzerland, Belgium, Norway and Spain | France, United Kingdom, Spain and Netherlands, predominance period July-October 2020. |
|
ΔFVI spike Cluster 5 |
Denmark (North Jutland) | First reports on the rise of COVID-19 cases on mink farms within Denmark commenced in early June, 2020. Denmark first reported the appearance of a variant of concern referred to as Cluster 5 on November 5, 2020. This mutation cluster had been detected from August to September, 2020. | Primarily Denmark with other mink-transmitted human SARS-CoV-2 mutated variant strains found in Lithuania, Netherlands, Spain, Sweden, Italy, Greece and the USA | Primarily Denmark, originally North Jutland, predominance period early June to the end of November, 2020. Affecting people who are involved in farming, culling and pelting of mink[27,41-43]. |
|
B.1.1.7 VoC 202012/01 UK Variant |
Southeast England with ongoing rapid dominance across Switzerland, Denmark and the USA | The emergence of a SARS-CoV-2 variant of Concern (VoC 202012/01, lineage B.1.1.7) during the month of November, 2020, in Southeast England was first reported in December, 2020[28,29]. | Global with ongoing changes in geographical frequencies | B.1.1.7 is considered to be up to 80% more transmissible, currently affecting more than 30 countries[30]. Reported increase in severity of the disease with suspected increase in mortality to be confirmed, particularly amongst older patients (70-84 and older)[30]. |
|
B.1.351 / K417N, E484K and N501Y 501.V2 / 20H/501Y.V2 (formerly 20C/501Y.V2) South African Variant |
South Africa (Nelson Mandela Bay) Eastern Cape Province | October 8, 2020[31] | At present, 66 countries have reported the presence of this variant. This variant is rapidly spreading into other countries[31]. | First detected at the beginning of October, 2020, it rapidly spread into Europe during December, 2020, affecting primarily France, Switzerland, Finland and the UK. Additional countries with high presence of B.1.351 include Sweden, The Netherlands, Israel, Germany, Belgium, Mayotte, USA, Austria and Zimbabwe. |
|
P.1 (descendent of B.1.1.28) VoC-202101/02 20J/501Y.V3 Brazilian Variant Gamma Variant |
Japan, Brazil (Manaus, State of Amazonas) | Circulation of P.1 variant appears to have commenced as early as mid-December (December 15-23), 2020, in the State of Amazonas, Brazil. On January 2, 2021, four travellers from Japan visited the Brazilian State of Amazonas, returning to Japan on January 6, 2021, on which date the Japanese National Institute of Infectious Diseases first detected this variant. The Brazilian variant was reported on January 12, 2021[32]. | The Brazilian variant has reached 31 countries as of March 18, 2021. | Most affected countries include Brazil and Italy, followed by Belgium, the USA, Peru and Germany. |
| B.1.427 and B.1.429 | USA West Coast (San Francisco, California) | B.1.427 and B.1.429 genomic sequences were identified by research laboratories in California during December, 2020, and February, 2021[33]. | USA West Coast | In May, 2020, these variants represented less than 1% of samples analysed, climbing to 54% by the end of February, 2021. |
|
B.1.526 E484K |
USA Northeast coast (New York City, New York) | First detected in New York city in November, 2020, this variant reached 27% prevalence by mid-February, 2021[34,35]. | USA Northeast coast | Since November, 2020, the B.1.526 / E484K variant has increased its prominence in the Northeast USA, particularly in New York City. As of March, 2021, it accounts for up to 45% of new cases. Fourteen states in the USA have been affected, with relevant incidence in Texas, Maryland and Wyoming. |
|
B.1.525 VUI-202102/03 E484K |
United Kingdom, Denmark and Nigeria | First reported on December 15, 2020[36], B.1.525 shares the E484K mutation present in B.1.351, P.1 and B.1.526, which appears to reduce immunity induced by naturally occurring infection or vaccination[8,9,37]. | Multiple countries affected with current changes in geographical frequencies. | United Kingdom, Denmark, Nigeria, USA and Germany |
|
B.1.617.1/G/452.V3 B.1.617.2 (Delta Variant) and B.1.617.3 |
India (Maharashtra) | B.1.617.1/G/452.V3 was first detected on October 5, 2020[44]. | Originally and primarily affecting India, the variant B.1.617 rapidly spread into the UK (February 22, 2021) and the USA (February 23, 2021). To date, global infectivity encompasses at least 34 countries. | Global with involvement of at least 34 countries, namely affecting India, UK, USA, Singapore, Germany, Australia, Denmark, Ireland, Italy and Belgium[44] |
| C.37 Lambda BS Variant | Peru | C.37 Lambda BS Variant was first detected in Lima Peru in late December, 2020[101]. | Global, primarily Chile, Argentina, Colombia, Ecuador, Mexico, USA, Canada, Germany and Israel. As of June 15, 2021, C.37 was considered a variant of interest by WHO[102]. | First reported in Lima, Peru, in late December, 2020, the C.37 Lambda BS variant of interest accounts for 97% of Peruvian SARS-CoV-2 genomic sequences since April, 2021. Also affecting other South American countries such as Argentina, Chile, Ecuador and Colombia, the Lambda variant has a main presence in 29 countries[102]. |
Table 1 Brief Outline of Early and Recent SARS-CoV-2 Global Variants of Interest and Concern
Although there are anecdotal reports dated from mid to late November, 2019, the earliest clinical documentation of individuals most likely infected with ancestral variants of SARS-CoV-2 is, as of to-date, December 8, 2019[1-6]. The first formal report of a new viral strain related to the onset of a severe acute respiratory syndrome documented as WH-Human 1 coronavirus dates from December 26, 2019[24]. However, the first in-depth analysis of ancestral SARS-CoV-2 variant types S (Serine) and L (Leucine) was the result of the characterisation of Single Nucleotide Variations (SNVs) found in the MT291826 strain of SARS-CoV-2 which, reported as early as December 30, 2019, represents one of the earliest strains affecting humans[23].
Table 1 provides details of some of the most recent SARS-CoV-2 variants of relevance and concern following the in-depth early characterisation of SARS-CoV-2 variant types S (Serine) and L (Leucine). To assist cross reference between current and early publications, in some cases, original names given to SARS-CoV-2 variants are provided in the first, left column in addition to current nomenclatures (Nextstrain and PANGO).
Analysis of the adaptive evolution of SARS-CoV-2 leading to increased infectivity and transmission has underlined the relevance of derived alleles of both synonymous and nonsynonymous mutations[21-23]. During the early stages of global SARS-CoV-2 infections, it appeared that there were a greater number of nonsynonymous mutations within the analysed samples, whereas synonymous mutations carried a greater frequency of derived mutations[22].
Unsurprisingly, the progressive numbers of beneficial mutations have taken place within the boundaries and junction between the S1 and S2 subunits of the Receptor Binding Domain (RBD) of the SARS-CoV-2 Spike protein, known to be one of the most variable regions of the coronavirus genome[2,21-24].
The resulting Single Nucleotide Variants (SNVs) of SARS-CoV-2 have lead, amongst other things, to an increased binding capacity to its Angiotensin Converting Enzyme 2 (ACE2) putative receptor, without affecting the RBD overall structure and sequence similarities shared between SARS-CoV-2 RBD and that of SARS-CoV RBD[38]. On the basis of these similarities, it has been proposed that increased binding capacity to ACE2 has been the result of convergent evolution between SARS-CoV-2 and SARS-CoV RBDs[38].
Chronologically, improved binding capacity of SARS-CoV-2 RBD to ACE2 due to beneficial mutations seems to have transitioned through the first emergence of S (Serine) Type into that of L (Leucine) Type, whereby a rapidly evolving SARS-CoV-2 variant with Spike (S protein) G614 replaced D614 as the more prevalent pandemic variant of SARS-CoV-2. Indeed, by mid-May, 2020, the G614 variant had spread to several countries as a result of the possible increased infectivity of SARS-CoV-2 G614 variant[39].
In parallel, due to the early lifting of travel restrictions across Europe during the summer of 2020, two new variants, namely 20A.EU1 and 20A.EU2, emerged as the most prevalent variants resulting from a period of quarantine-free mobility amongst numerous European countries, underlining the importance of keeping and reinforcing quarantine procedures even during a greater exposure of SARS-CoV-2 to UV light in the course of prolonged, sunny summer days[40]. These two variants remained amongst the most relevant in Europe from June to October of 2020, with different frequencies in Western European countries[40].
In one of our earlier reports related to a dynamic multifactorial model on the origins of SARS-CoV-2[4], we emphasised the direct impact of intentional and otherwise accidental anthropogenic activities leading to both interspecies (animals-to-humans-to-animals) and intraspecies (humans-to-humans, animals-to-animals) infectivity and transmission of current and ancestral SARS-CoV-2 variants[4].
Early indications of interspecies SARS-CoV-2 transmission from humans to domesticated animals such as cats and ferrets, with subsequent intraspecies infectivity involving cats-to-cats and ferrets-to-ferrets transmission and infectivity[4], were made more evident by the recent emergence of the ΔFVI spike/Cluster 5 variant (see Table 1).
First transmitted from infected mink farmers to mink animals, acting as a SARS-CoV-2 potential virus reservoir, farmed mink seemed to have accelerated the emergence of the new ΔFVI spike/Cluster 5 variant, carrying three substitutions and one deletion in the spike protein[27,41-43].
These genetic changes conferred the ΔFVI spike/Cluster 5 variant an increased transmission and infectivity capacity, rapidly spreading into other mink farms across Europe, primarily within Italy, Spain and Sweden, as well as the United States in North America[27,41-43].
The ΔFVI spike/Cluster 5 variant lasted from June, 2020, to the end of November, 2020, with no further detection after the culling of more than 17 million mink animals, the destruction of their raw pelts, the quarantine of mink workers and the closure of mink farms and related facilities[27,41-43].
The rapid interspecies transmission of human infections to highly secluded animals such as mink highlights the pressing need to halt the use of raw pelts obtained from live animals instead of man-made, durable and highly performant materials currently available for tailoring every day and luxurious garments.
Most likely reaching 200 million infections globally by the end of July, 2021, the COVID-19 pandemic outbreak has been sustained not solely by the relatively high mutation rates of SARS-CoV-2 types, equivalent to ≥ 6.0 x 10-6 nucleotide substitutions per site daily or 0.2 mutations per genome per day, but also by an array of advantageous mutations.
Most of the current SARS-CoV-2 beneficial mutations have taken place at the S1/S2 junction and boundaries between the S1 and S2 subunits of the Receptor Binding Domain (RBD) of the SARS-CoV-2 Spike protein, known to be one of the most variable regions of the coronavirus genome[2,21-24]. These mutations comprise, among others[2-4,21-24]:
- Synonymous / Nonsynonymous
- Missense
- Deletions / Insertions
- Frameshift deletions
- Stop-gained
- SARS-CoV-2 Variants of Interest B.1.526, B.1.526.1, B.1.525, P.2, B.1.617, B.1.617.1, B.1.617.2, B.1.617.3
- SARS-CoV-2 Variants of Concern B.1.1.7, P.1, B.1.351, B.1.427, B.1.429
- SARS-CoV-2 Variants of High Consequence (no SARS-CoV-2 variants with high consequence have been detected as of to date)
- Operation Warp Speed (OWS) - Under the auspices and coordination of the USA Health and Human Services (HHS) and the private sector, OWS made possible the development, industrial manufacturing, regulatory approval and deployment of some of the major vaccines currently in use such as Moderna/NIH mRNA-1273, Johnson & Johnson JNJ-78436735, AstraZeneca/University of Oxford AZD1222 and Novavax NVX-CoV2373, amongst several other candidate vaccines such as Merk/Themis V591 and Sanofi-GSK VAT00008.
- Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) - Coordinated by the Foundation of the National Institutes of Health (FNIH) in collaboration with NIH and at least 16 Biopharmaceutical Corporations, ACTIV aims to fast-track the standardisation and sharing of pre-clinical evaluation methodologies, to prioritise and accelerate the clinical evaluation of biotherapeutic candidates, to optimise clinical trial capacity and to advance vaccine development.
- The COVID-19 Prevention Trials Network (COVPN) - The National Institute of Allergy and Infectious Diseases (NIAID) established a selective clinical trial network to enable thousands of volunteers to participate in clinical trials to assess the safety and efficacy of investigational vaccines and monoclonal antibodies for the prevention of COVID-19.
- COVID-19 Vaccines Global Access (COVAX) - Directed and coordinated by the Vaccine Alliance (Gavi), the Coalition for Epidemic Preparedness Innovations (CEPI) and WHO, COVAX has articulated strategies with at least 10 Biopharmaceutical Companies to secure low-cost COVID-19 vaccines available to all countries, amongst other international efforts aimed at curbing the ongoing COVID-19 outbreak including supporting the use of combined vaccination strategies involving more than one vaccine type.
- Total Serious / Non-Serious Rate Ratio
- Moderna/NIH (0.09)
- Pfizer/BioNTech (0.35)
- Oxford/AstraZeneca (0.44)
- Protection against current variants of concern
- Pfizer/BioNTech
- Moderna/NIH
- Oxford/AstraZeneca
- Bat coronavirus Rm YN02
- Bat coronavirus RaTG13
- SARS-CoV-2-Related Lineage Serine S Type SNP 28,144:UCC ORF8
- SARS-CoV-2-Related Lineage Leucine L Type SNP 8,782:CUU orf1ab
- WH-Human 1 coronavirus
- MT291826
- Total Serious / Non-Serious Rate Ratio
- Moderna/NIH (0.09)
- Pfizer/BioNTech (0.35)
- Oxford/AstraZeneca (0.44)
- Protection against current variants of concern
- Pfizer/BioNTech
- Moderna/NIH
- Oxford/AstraZeneca
Although the relative reported number and asymmetrical geographical distribution of thousands of emerging SARS-CoV-2 variants will be described below, to date, current emerging variants have been associated with three different distinct classes / categories in accordance with their level of threat to global human health[45]. These classes comprise:
The asymmetric emergence of SARS-CoV-2 variants with various degrees of infectivity and transmission are the result of several beneficial mutations enabling, amongst other things, an increased fitness through ligand / receptor affinity modulation and the processing of viral / receptor complexes, particularly during immediate viral early entry to permissive cells, all assumed to carry the putative ACE2 receptor[1-12,20-23,25-30,37-40,46,47].
From the perspective of global health, it is clear that our early estimate of a fairly high mutation rate predicted for SARS-CoV-2[4] has already lead to immune escape. Disquieting evidence for convergent evolution is equally reflected in mutations encompassing N501Y and E484K, present in several variants of interest / concern such as B.1.526, B1.525 and P.3[28].
In this regard, it is important to emphasise that the classification of emerging variants, cited above, into variants of interest and variants of concern can change as more current available data provide a better picture in terms of the implications to public health of specific well-established and emerging variants.
The implications of more current mutations, involving variants B.1.617.1, B.1.617.2 and B.1.617.3, as well as the N-Terminal Domain of the SARS-CoV-2 Spike peptide, are covered in other sections.
From the Classification of SARS-CoV-2 Genome into Six Viral Types to Evidence of Viral Adaptive Episodes by Clonal Interference and Molecular Convergent Evolution: Implications for Public Health Measures and Genomic Surveillance
In spite of the rapid and relentless speed with which the SARS-CoV-2 outbreak spread from its initial onset in China to the rest of the world, the identification and genomic characterisation of the ancestral strains of SARS-CoV-2 leading to the ongoing COVID-19 pandemic outbreak have also been achieved within unparalleled time frames.
Based upon the original SARS-CoV-2 viral samples obtained from some of the first patients affected by COVID-19, a reference sequence, namely the 29,903 nucleotides WH-Human 1 / Wuhan-Hu-1 strain originally isolated in China[24] has enabled the identification of Single Nucleotide Variations (SNVs) present in subsequent genomic analysis of clinical samples obtained from a larger population of infected individuals[2,6,21].
Recent genomic analyses based upon single nucleotide variations (SNVs), performed in tandem using approximately 2, 6 and 40 x 103 complete SARS-CoV-2 genome sequence analysis within protein coding and 5' untranslated viral regions, were used to define signature variation clusters leading to the classification of SARS-CoV-2 into six viral strain types[21]. Table 2 summarises the main genomic characteristic of Types I-VI, including subtypes VIa and VIb.
Amongst some of the most relevant findings of tandem clustering analyses is the presence of 13 signature SNVs within SARS-CoV-2 protein coding regions and one SNV in the 5' untranslated region (UTR), some of which encompassed strong allelic associations[21]. This is the case, for instance, for signature SNVs C8782T and C28144C, known to possess a high coefficient of allelic association of R2 = 0.987[21].
Interestingly, both SNVs first co-appeared in strain MT291826, one of the first isolates of SARS-CoV-2, reported as early as December 30, 2019, only a few days after the identification and characterisation of the putative WH-Human 1 / Wuhan-Hu-1 coronavirus found in some of the first clinical samples obtained during the early onset of COVID-19 in Wuhan, China[24].
The extraordinarily high allelic association shared between SNVs C8782T and C28144C is of great interest to the better understanding of the early adaptive evolution of SARS-CoV-2. Based upon their strong allelic association, it is highly likely that these early SNVs were initially co-transmitted during early stages of SARS-CoV-2 human infections, both being actually used to designate the S (Serine) and L (Leucine) Types of SARS-CoV-2, whereas the S (Serine) Type classified within the viral strain Type II is not only closer to bat SARSr-CoV RaTG13 but is most likely related to the original strain of SARS-CoV-2 infecting humans, following concurrent asymmetrical patterns of transmission[4,21,22].
The co-transmission and asymmetrical co-existence of several viral strains suggests not only fairly rapid episodes of viral adaptive evolution through the variety of mutations types previously outlined but also the capacity of SARS-CoV-2 to increase fitness by enhancing, amongst other things, the binding capability of the Spike protein to its putative ACE2 receptor whilst equally reducing antibody neutralisation[4,6-12,46,47].
| Viral Strain Type | nt1059 (nsp2) T265I | nt1397 (nsp2) V378I | nt3037 (nsp3) F924F | nt8782 (nsp4) S2839S | nt11083 (nsp6) L3606F | nt14408 (nsp12) P4715L | nt14805 (nsp12) Y4847Y | nt17747 (nsp13) P5828L | nt17858 (nsp13) Y5865C |
| Type-I | T | V | F | S | L | P | Y | P | Y |
| Type-II | S | ||||||||
| Type-III | F | Y | |||||||
| Type-IV | S | L | C | ||||||
| Type-V | I | F | |||||||
| Type-VI | F | L | |||||||
| Type-VIa | I | F | L | ||||||
| Type-VIb | F | L |
| Viral Strain Type | nt18060 (nsp14) L5932L | nt23403 (S) D614G | nt25563 (ORF3a) Q57H | nt26144 (ORF3a) G251V | nt28144 (ORF8) L84S | nt28688 (N) L139L | nt28881 (N) R203K | nt28882 (N) R203K | nt28883 (N) G204R |
| Type-I | L | D | Q | G | L | L | R | R | G |
| Type-II | S | ||||||||
| Type-III | Y | ||||||||
| Type-IV | L | S | |||||||
| Type-V | L | ||||||||
| Type-VI | F | G | |||||||
| Type-VIa | I | G | H | ||||||
| Type-VIb | G | K | K | R |
| Viral Strain Type | nt241 (5'UTR) | nt1059 (nsp2) | nt1397 (nsp2) | nt3037 (nsp3) | nt8782 (nsp4) | nt11083 (nsp6) | nt14408 (nsp12) | nt14805 (nsp12) | nt17747 (nsp13) | Nt17858 (nsp13) |
| Type-I | C | C | G | C | C | G | C | C | C | A |
| Type-II | T | |||||||||
| Type-III | T | T | ||||||||
| Type-IV | T | T | G | |||||||
| Type-V | A | T | ||||||||
| Type-VI | T | T | T | |||||||
| Type-VIa | T | T | T | T | ||||||
| Type-VIb | T | T | T |
| Viral Strain Type | nt18060 (nsp14) | nt23403 (S) | nt25563 (ORF3a) | nt26144 (ORF3a) | nt28144 (ORF8) | nt28688 (N) | nt28881 (N) | nt28882 (N) | nt28883 (N) |
| Type-I | C | A | G | G | T | T | G | G | G |
| Type-II | C | ||||||||
| Type-III | T | ||||||||
| Type-IV | T | C | |||||||
| Type-V | C | ||||||||
| Type-VI | G | ||||||||
| Type-VIa | G | T | |||||||
| Type-VIb | G | A | A | C |
Table 2 Classification of SARS-CoV-2 Viral Strains based on Single Nucleotide (SNVs) Variation Analysis
Genomic analysis based upon single nucleotide variations (SNVs) performed in tandem using approximately 2, 6 and 40 x 103 complete SARS-CoV-2 genome sequence analysis within protein coding and 5' untranslated viral regions was used to define signature variation clusters leading to the classification of SARS-CoV-2 into six distinct viral strain types[21]. Variations are shown as single letter amino acids below their corresponding viral genome positions. Variants types are characterised, amongst other things, by the presence of both synonymous and nonsynonymous mutations when compared to the 29,903 nucleotides strain WH-Human 1 / Wuhan-Hu-1 originally isolated in China and used as the reference genome[24]. With the exception of Type I SARS-CoV-2 genomes, which carry only one or none of the 13 signature SNVs, each of the remaining Types [II-VI (VIa-VIb)] were defined with at least two out of the 13 signature SNVs within SARS-CoV-2 protein coding regions[21].
Prior to the articulation of consistent and more encompassing global public health interventions, potential co-transmission of SNVs possessing high allelic associations could have enabled early SARS-CoV-2 variants to gain dominance through clonal interference, whereby at least more than two beneficial mutations achieved simultaneous population fixation during the course of independent asymmetric evolution through enhanced transmission and infectivity similar to that previously reported in other RNA viruses[15].
Evidence in support of SARS-CoV-2 evolution through clonal interference involves, amongst other things, the co-existence of SARS-CoV-2 S (Serine) and L (Leucine) Types as well as Type II and Type VI viral strain types, whereby the ancestral SARS-CoV-2 S (Serine) and the Type II viral strains were displaced by the L (Leucine) and Type VI SARS-CoV-2 strains, respectively[21,22].
In addition to clonal interference, the combined effect of close to 200 million infections globally, together with relatively high mutation rates for SARS-CoV-2 types being equivalent to ≥ 6.0 x 10-6 nucleotide substitutions per site daily or 0.2 mutations per genome per day[4], have led to the co-occurrence of more than two viral variants that appear to have reduced viral susceptibility to vaccine-induced humoral immunity and response to neutralising polyclonal antibodies[48].
Such a level of combined SARS-CoV-2 adaptive fitness capable of evading immune response has raised the concern of the potential occurrence of convergent evolution currently encompassing mutations such as N501Y and E484K, present in several variants of interest / concern such as B.1.526, B1.525 and P.3[28].
The rather diverse and versatile mechanisms of adaptive evolution of SARS-CoV-2's current and emerging variants, encompassing clonal interference and convergent evolution, calls for more consistent and robust public health measures including routine global genomic surveillance as well as the testing of clinical samples from patients living in regions with the known presence of more than one variant of interest / concern, searching for possible co-infections in COVID-19 patients. Table 3 summarises current global efforts and voluntary reporting of genomic surveillance to the Nextstrain database, describing lineages and emerging variants with the support of the GISAID Initiative.
According to data publicly available as of May 27, 2021, only 38 countries, representing merely 17% of all countries affected by COVID-19, have performed and shared genomic analyses related to the emergence of SARS-CoV-2 mutated genomes. In addition to the small number of participating countries, it appears that the amount of information shared by each country is not directly related to its individual available resources, technical capability and infrastructure.
| Country | Reported Number of Variants | Reported Number of Variants / Total Population (millions)* | Variant Incidence Rates %** | Estimated Number of Positive Cases Carrying a New Variant** |
| Australia | 37 | 1.46 | 0.13 | 3,803.15 |
| Belgium | 9 | 0.78 | 0.001 | 866.06 |
| Brazil | 15 | 0.07 | 0.0001 | 1,249.036 |
| Cambodia | 2 | 0.12 | 0.09 | 200.97 |
| Canada | 20 | 0.53 | 0.002 | 1,922.17 |
| Chile | 7 | 0.37 | 0.0007 | 684.07 |
| China | 323 | 0.22 | 0.36 | 32,460.12 |
| Congo | 1 | 0.01 | 0.01 | 96.81 |
| Czech Republic | 2 | 0.18 | 0.0001 | 151.5 |
| Denmark | 2 | 0.34 | 0.0009 | 205.21 |
| Finland | 19 | 3.44 | 0.025 | 1,900.08 |
| France | 39 | 0.58 | 0.0009 | 4,057.72 |
| Georgia | 4 | 1.08 | 0.0014 | 446.15 |
| Germany | 30 | 0.36 | 0.001 | 2,776 |
| Hong Kong | 24 | 3.2 | 0.21 | 2,403.87 |
| India | 4 | 0.003 | 0.000033 | 395.6 |
| Italy | 10 | 0.17 | 0.0003 | 1,059.62 |
| Ireland | 6 | 1.22 | 0.003 | 701.81 |
| Japan | 16 | 0.13 | 0.003 | 1,400.55 |
| Luxembourg | 2 | 3.2 | 0.003 | 182.27 |
| Mexico | 2 | 0.015 | 0.00009 | 200.23 |
| Nepal | 2 | 0.07 | 0.0007 | 193.79 |
| Netherlands | 290 | 16.78 | 0.023 | 28,806.051 |
| New Zealand | 1 | 0.2 | 0.04 | 99.28 |
| Nigeria | 2 | 0.001 | 0.001 | 162.49 |
| Northern Ireland | 1 | 0.53 | 0.0009 | 105.3 |
| Panama | 1 | 0.235 | 0.0003 | 106.05 |
| Portugal | 4 | 0.39 | 0.0005 | 410.2 |
| Russia | 1 | 0.007 | 0.00002 | 90.4 |
| Singapore | 28 | 4.9 | 0.05 | 3,015 |
| Spain | 5 | 0.106 | 0.0002 | 651.06 |
| South Korea | 24 | 0.464 | 0.024 | 2,442.168 |
| Switzerland | 45 | 5.27 | 0.008 | 4,737.74 |
| Taiwan | 15 | 0.64 | 1.47 | 1,502.34 |
| Thailand | 4 | 0.057 | 0.014 | 402.276 |
| United Kingdom | 111 | 1.67 | 0.003 | 12,999.126 |
| USA | 169 | 0.514 | 0.0005 | 15,461.18 |
| Vietnam | 2 | 0.021 | 0.08 | 207.28 |
| Total Reported | 1279 |
Table 3 Global Voluntary Reporting of Emerging SARS-CoV-2 Genomes
As of May 27, 2021, only 38 out of 222 countries known to have active daily SARS-CoV-2 infections have performed and contributed to global genomic analysis unveiling emerging mutated SARS-CoV-2 genomes through a concerted voluntary reporting system. This involves genomic surveillance and reporting to the Nextstrain database describing lineages and emerging variants with the support of the GISAID Initiative. There are three distinct groups with estimated numbers of SARS-CoV-2 positive cases carrying a new variant ranging as follows: Group 1 12,000-35,000 (Dark Purple); Group 2 1,000-5,000 (Medium Dark Purple); Group 3 90-1000 (Light Purple). Whilst countries included within each group clearly reflect an asymmetrical distribution of emerging SARS-CoV-2 mutated genomes, the relevance of the actual variant incidence rate values, based upon estimated COVID-19 cases reported in March, 2021, remains relative due to the actual technical capabilities of each country as well as its willingness to voluntary contribute to available public databases. Although of relative actual value, the combined variant incidence rates and estimated number of cases carrying a new variant in countries such as Taiwan underline the usefulness of routine genomic analysis and surveillance to effectively abrogate transmission and infectivity of COVID-19.
* Based upon Estimated Total Country Population Reported in 2019
** Based upon Estimated COVID-19 Cases Reported in March 2021
Participating institutions from 38 countries provide a glimpse into the asymmetric emergence and genomic evolution of SARS-CoV-2, allowing us to distinguish three major groups based upon the estimated number of SARS-CoV-2 positive cases carrying a new variant. The ranges of estimated SARS-CoV-2 positive cases carrying a new variant and the countries belonging to each group are shown in Table 4.
Due to several concurrent factors, such as regional testing capabilities and reporting, and different public health measures, including vaccination rates by country (as of July 18, 2021, merely 12.97% of the global population reports being fully vaccinated), it is important to increase global SARS-CoV-2 genomic surveillance and centralised accessible data reporting across at least 75% of all countries affected by COVID-19.
Reliable SARS-CoV-2 genomic surveillance and fast information sharing have been central components of effective COVID-19 disease control in countries, such as Taiwan, that have embraced this practice, together with additional public health measures, since the beginning of the ongoing pandemic outbreak.
From a broader perspective, consistent and routine SARS-CoV-2 genomic analysis and surveillance will continue to not only assist us in better understanding the genomic adaptive evolution of SARS-CoV-2, through processes such as clonal interference and molecular convergent evolution of selective beneficial mutations, but to continue making changes in the formulation of the new generation of highly effective RNA vaccines which, regardless of their high degree of novelty, continue to be the safest and most highly effective vaccines amongst all vaccines currently being used, worldwide.
|
Group 1 12,000-35,000 Cases |
Group 2 1,000-5,000 Cases |
Group 3 90-1000 Cases |
|
China Netherlands United Kingdom United States |
Australia Brazil Canada Finland France Germany Hong Kong Italy Japan Singapore South Korea Switzerland Taiwan |
Belgium Cambodia Chile Congo Czech Republic Denmark Georgia India Ireland Luxembourg Mexico Nepal New Zealand Nigeria Northern Ireland Panama Portugal Russia Spain Thailand Vietnam |
Table 4 Asymmetric emergence and genomic evolution of SARS-CoV-2
Although the estimated number of positive cases carrying a new genomic mutation is not an absolute number, it provides evidence of an asymmetric emergence and evolution of SARS-CoV-2. It is worth noting that countries, such as Taiwan, which embraced early SARS-CoV-2 genomic surveillance as a central part of their disease outbreak control and contact tracing capabilities amongst other early and effective public health measures have been at the forefront of minimising the impact of the ongoing COVID-19 outbreak.
Robust, routine and highly accessible global genomic characterisation and surveillance is essential to rapid intervention in the unforeseeable event of the emergence of SARS-CoV-2 super-variants that could lead to high-consequence public health concerns including increased disease severity, infectivity, hospitalisation and global death rates.
This activity is equally essential to better understand, further diagnose and treat current survivors of COVID-19, patients who continue to suffer from a variety of lingering symptoms and neurological disorders that include dizziness, fatigue, brain fog, headaches, chest and muscular pains as well as damage to several organs recently identified as Post-Acute COVI-19 syndrome[49].
There are several other practical uses of Global Voluntary Reporting of Emerging SARS-CoV-2 Genomes. As shown in Figure 1, based upon the data summarised in Table 3, rapidly produced graphical representations of the actual geographical distribution of genomic variants (Figure 1A) and the estimated number of COVID-19 patients potentially carrying a new variant (Figure 1B) can be used to better pinpoint regions more affected by the pandemic, taking into account that detection and reporting is evenly performed.
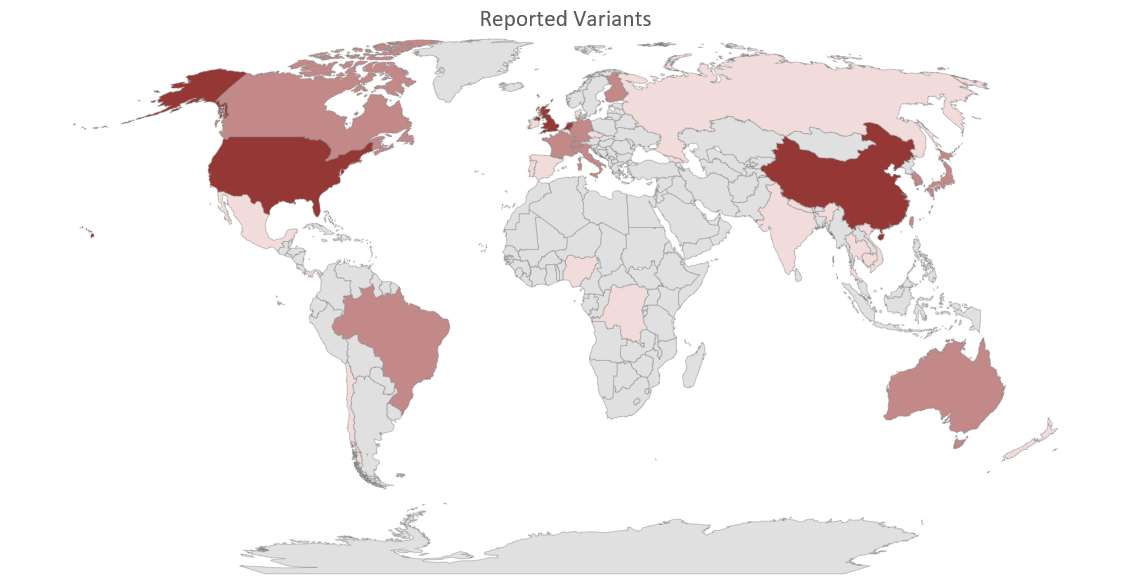
Figure 1A Geographical Location of Newly Reported Genomic Variants as of May 27, 2021
There are three distinct groups, with estimated number of SARS-CoV-2 positive cases carrying a new variant ranging as follows: Group 1 12,000-35,000 (Dark Purple); Group 2 1,000-5,000 (Medium Dark Purple); Group 3 90-1000 (Light Purple). Details of each group are presented in Table 4, above, and Figure 1B, below.
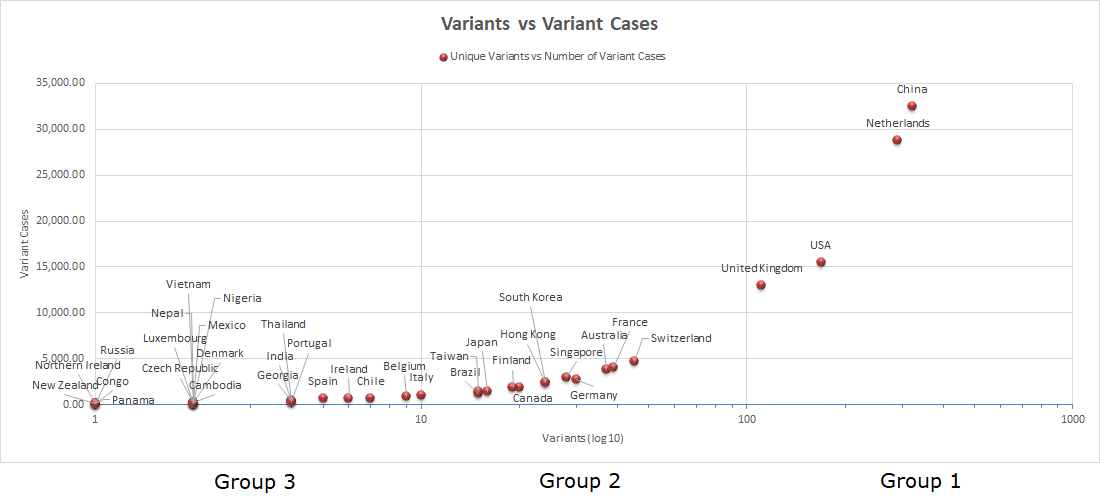
Figure 1B Asymmetric Distribution of Unique Variants vs Number of Variant Cases
Although global centralised and routine SARS-CoV-2 genomic analysis performed in confirmed COVID-19 cases is still largely unavailable, current shared data concerning emerging mutated SARS-CoV-2 genomes demonstrate the utmost relevance of performing genomic surveillance in order to better predict the geographical location and genomic characteristics of emerging variants of interest / concern as well as their potential co-transmission.
The asymmetric distribution of emerging genomic variants, based upon data voluntary reported by 38 countries, is highly indicative of the immediate need to increase global SARS-CoV-2 genomic surveillance and, in some cases, proceed with the isolation and detailed characterisation of the biological properties of viral variants carrying more than one mutation of interest.
This activity is not only vital to the continued assessment of the adaptive evolution of SARS-CoV-2 as we continue expanding global vaccination campaigns but is also critical to the ability to rapidly adapt current vaccines in order to increase their safety and efficacy as well as to enable better assessment of the real risks associated to the potential emergence of SARS-CoV-2 super-variants.
Current Phylogenetic Analysis of SARS-CoV-2 Genome and Possible Ancestral Origins
Beyond the obvious biological relevance of finding the phylogeny and evolutionary adaptations leading to the origins of SARS-CoV-2, the identification and genomic characterisation of the closest known viral intermediates is pivotal to articulating effective public preventative health measures, including the design of safe and effective vaccines capable of eliciting highly selective and enduring immune responses.
In previous reports, based upon the substantial level of sequence homology shared between SARS-CoV-2 and bat SARSr-CoV RaTG13 (96.2%) as well as the potential common capacity of the Spike Receptor Binding Domains (RBDs) of SARS-CoV-2 and bat SARSr-CoV RaTG13 to elicit binding to the putative Angiotensin Converting Enzyme 2 (ACE2) receptor, we advanced the concept of a direct transmission of an ancestral bat SARSr-CoVRaTG13 strain from bats to humans[4].
According to our multifactorial model on the origins of SARS-CoV-2, the adaptive genomic changes to an ancestral SARSr-CoVRaTG13 precursor required for successful viral infectivity, replication and transmission could have been achieved through human-to-human super-spreader-mediated transmission initially leading to an attenuated level of infection[4].
An initial attenuated level of infection could have set the stage for a progressive genomic evolution in humans, giving rise to the first successfully infectious SARS-CoV-2 viral strains isolated from five different patients at very early stages of the SARS-CoV-2/COVID-19 pandemic outbreak sharing 99.9% of sequence homology[1-6]. Due to their high level of sequence homology (99.9%) and their isolation and characterisation at very early stages of the emergence of COVID-19, these strains hold the key to further refining the origins of SARS-CoV-2.
Recent and substantial contributions have brought us closer to further deciphering the origins and evolution of SARS-CoV-2. One of the most salient characteristics of SARS-CoV-2 relates to the presence of a polybasic Furin (S-Pro-Arg-Arg-Ala-R) cleavage site motif that, being absent in any coronavirus of the same clade, including bat SARSr-CoV RaTG13, is believed to be involved in the effective transmission and infectivity of SARS-CoV-2[1,4,11-13].
Until recently, the absence of the polybasic Furin (S-Pro-Arg-Arg-Ala-R/PRRA) cleavage site motif in bat SARSr-CoV RaTG13 and other coronaviruses of the same clade had limited our ability to fully support our model in which a direct primordial infection of an ancestral SARSr-CoVRaTG13 from bats to humans followed by human adaptive evolution through human-to-human super-spreader-mediated transmission could have led to the emergence of the ancestral S (Serine) / Type II variants of SARS-CoV-2[4].
However, the generation of a polybasic Furin mutant lacking the Furin (S-Pro-Arg-Arg-Ala-R/PRRA) cleavage site motif (ΔPRRA) using a SARS-CoV-2 reverse genetic system demonstrated the ability of ΔPRRA to elicit attenuated infections both in human cells, including Calu-3 human lung cancer cell lines, and in animal models[14].
Such an attenuated infection by the SARS-CoV-2 ΔPRRA mutant not only supports our model involving a possible direct primordial infection of an ancestral SARSr-CoVRaTG13 from bats to humans, it also signals the presence of immediate viral entry accessory receptors (IVEARs) that could explain the efficient replication of SARS-CoV-2 found in a variety of permissive cells[50].
In this regard, the relevance of Leucine Aminopeptidase (LAPase) and Neuropilin-1 in the pathophysiology of SARS-CoV-2, including the development of post-acute COVID-19 syndrome, is discussed below.
The extensive recent analysis of full-length genomic sequences of several sarbecoviruses, including SARS-CoV-2 closely related viruses in bats and pangolins, has led to the construction of a detailed phylogenetic tree using the neighbour joining method, elucidating the closest SARS-CoV-2 coronaviruses to date[13]. A contemporary adaptation of sarbecoviruses phylogenetic tree depicting the closest and ancestral types of SARS-CoV-2 is shown in Figures 2A and 2B.
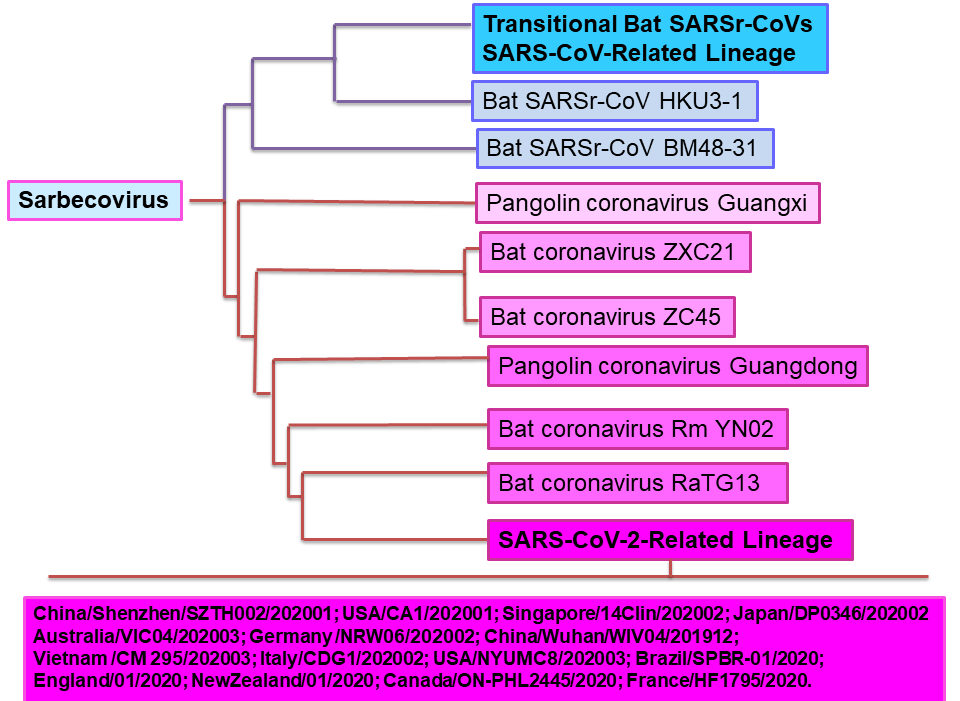
Figure 2A Partial Sarbecovirus Phylogenetic Tree with location of SARS-CoV-2-Related Lineage
Using genomic sequences available from GISAID and GenBank databases as of October, 2020, including closely related viruses in bats and pangolins, a phylogenetic tree was constructed by the joining neighbour method[13]. Figure 2A shows the pangolin and bat coronaviruses with known full genomic sequences closest to SARS-CoV-2. The most recent SARS-CoV-2 related lineage is also shown, including only some of the most current genomic variants.
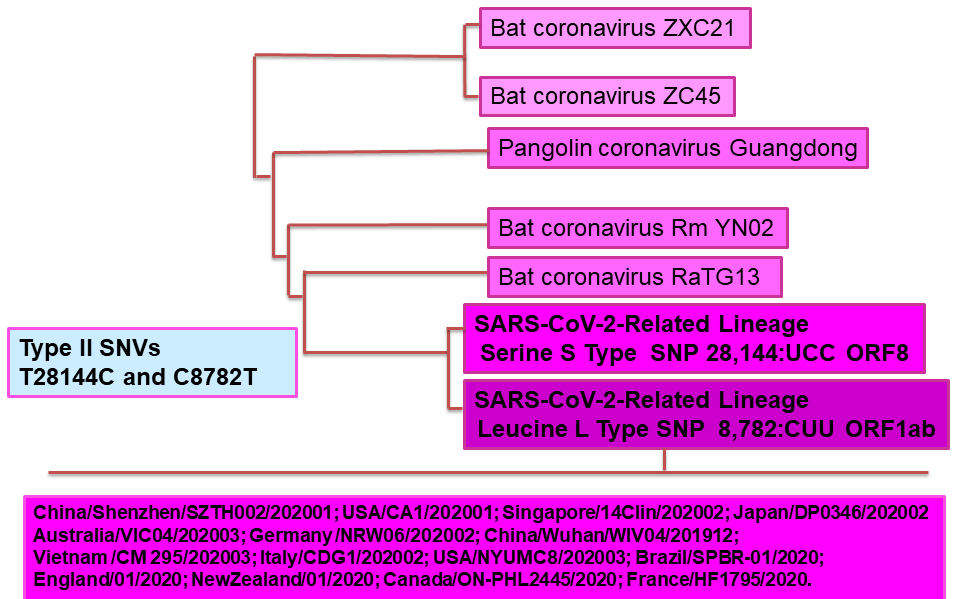
Figure 2B Partial Sarbecovirus Phylogenetic Tree with some of the closest SARS-CoV-2 Genomic Sequences
Recent Signature variation clusters elucidated from single nucleotide variations (SNVs) performed in tandem using approximately 2, 6 and 40 x 103 complete SARS-CoV-2 genome sequence analyses in protein coding and 5' untranslated regions have unveiled six distinct strain types (Type I-VI [VIa,b])[21]. As shown in Figure 2B, Type II contains SNVs C8782T (Serine) and T28144C (Leucine), which were also used to define the S (Serine) and L (Leucine) type of SARS-CoV-2, respectively, equally encompassing their corresponding non-synonymous point mutations C28144 (Serine) / T28144 (Leucine)[22]. Present in the strain MT291826 of SARS-CoV-2, they represent some of the earliest genomic characteristics of SARS-CoV-2[4,21-23].
Perhaps, one of the most relevant findings emerging from extensive genomic analysis of SARS-CoV-2 has been the identification of the Single Nucleotide Variants (SNVs) C8782T (Serine) and C28144C (Serine). Remarkably, both SNVs seem to have first co-appeared in strain MT291826 of SARS-CoV-2[21]. Identified and reported as early as December 30, 2019, merely four days after the original isolation of WH-Human 1 coronavirus on December 26, 2019, MT291826 represents one of the earliest SARS-CoV-2 variant strains found in China, belonging to Clade L84S[21-23].
Additionally, as mentioned earlier, SNVs C8782T (Serine) and C28144C (Serine) possess a strong coefficient of allelic association of R2 = 0.987[21]. Hence, it is plausible that these early SNVs were co-transmitted in the course of initial and subsequent infections, most likely with concurrent or sequential incidences leading to in vivo asymmetrical mutation patterns of ancestral forms of SARS-CoV-2 that were, most likely, close to the S (Serine) ancestral Type[4,21,22].
Comparative analysis of SNVs within Type II variants, including the S (Serine) Type of SARS-CoV-2 against those found in various animal coronaviruses, strongly suggests that the S (Serine) Type is not only closer to bat SARSr-CoV RaTG13 but is, most likely, closer to the original strain of SARS-CoV-2 infecting humans[4,21,22]. Figure 2B illustrates a partial sarbecovirus phylogenetic tree with the closest SARS-CoV-2 genomic sequences.
In our previous model of the multifactorial origins of SARS-CoV-2, we addressed both salient natural events that could have taken place leading to the zoonotic transmission of SARS-CoV-2 from bats to humans as well as an otherwise accidental transmission during the tailoring of selective coronavirus-based vectors potentially used for the design of what could have been a universal vaccine against HIV, Malaria and, perhaps, other pathogens of current global health concern[4].
Although the precise characterisation of the origins of SARS-CoV-2 will remain difficult to establish due, amongst other things, to the fairly rapid closure and sanitisation of all possible affected areas within the province of Hubei where the first clinical cases of COVID-19 were reported, based upon some of the main characteristics of the sabercovirus phylogenetic tree (partially summarised in Figures 2A and 2B), the following set of experiments could provide additional insight into the potential origins and adaptive evolution of SARS-CoV-2 viral ancestral genomes.
In a comparative study, evaluate in vitro replication and cytopathicity in Vero E6 and Calu-3 2B4 cells at low multiplicity of infection (0.01 plaque-forming units [PFU] per cell) of some of the viruses included in Figure 2B, such as Bat coronavirus Rm YN02, Bat coronavirus RaTG13, SARS-CoV-2-Related Lineage Serine S Type SNP 28,144:UCC ORF8, SARS-CoV-2-Related Lineage Leucine L Type SNP 8,782:CUU orf1ab, WH-Human 1 coronavirus and MT291826. It would be equally useful to perform a second set of similar in vivo studies using the hamster model of SARS-CoV-2 pathogenesis[51].
Considering the potential early co-transmission of SNVs C8782T (Serine) and C28144C (Serine) found in the otherwise ancestral S (Serine) Type, it would be equally useful to evaluate the replication and cytopathicity of single mutations involving SNV C8782T alone, SNV C28144C alone as well as that in which both potentially co-transmitted SNVs are removed.
Immediate Viral Entry Accessory Receptors (IVEARs): the Anticipated Role of Leucine Aminopeptidase
Two separate findings have shaped our recent understanding of the mechanisms involved in SARS-CoV-2 viral entry. The first is the recent discovery of the role of Neuropilin-1 as an accessory host receptor for SARS-CoV-2 with potential implications in some of the neuropathological disorders experienced by COVID-19 patients[52-56].
The second encompasses the ability of the SARS-CoV-2 polybasic Furin mutant ΔPRRA to replicate and sustain an attenuated level of infectivity conferring protection against re-challenge with the parental SARS-CoV-2 variant[14]. Although these independent findings were initially intended to further understand, on the one hand, the relevance of the polybasic Furin cleavage motif within the Spike protein and, on the other hand, the neurological implications induced by SARS-CoV-2 infectivity, there appears to be an underlining consequential molecular pathway triggered during SARS-CoV-2 cell surface viral processing and immediate viral entry events.
During the course of these exquisitely coordinated events, following cleavage of the SARS-CoV-2 Spike protein at the S1/S2 Furin junction of the Receptor Binding Domain (RBD), the resulting putative S1 CendR motif acts as an anchoring ligand to cell surface Neuropilin-1 molecules[52].
Although the exact intracellular ramifications of these sequential events taking place during immediate viral entry remain to be elucidated, they are thought to play a role in the endocytic pathway of SARS-CoV-2 because Neuropilins have been found to facilitate the internalisation of CendR ligands by means of endocytic processes reminiscent of micropinocytosis[52].
These sequential events provided us with clear evidence that, in addition to Neuropilin-1, other immediate viral entry accessory receptors (IVEARs) could be involved. This concept is supported by the fact that the SARS-CoV-2 polybasic Furin mutant ΔPRRA can replicate and sustain an attenuated level of infectivity despite having hindered the sequential release of S1 CendR motif required for SARS-CoV-2 binding and internalisation through the Neuropilin-1-dependent pathway due to the removal of the Furin cleavage from the S1/S2 junction of SARS-CoV-2 RBD[14,52].
In addition to the dual role of cellular enzymes such as Angiotensin Converting Enzyme 2 (ACE2) implicated in controlling blood pressure through degradation of Angiotensin II as well as serving as anchoring receptors for RNA viruses such as some coronaviruses including SARS-CoV-2, selective aminopeptidases belonging to the M1 family have been shown to play similar roles[17-19,57-58].
In particular, we have provided ample evidence that Leucine Aminopetidase (LAPase) can potentiate HIV infectivity during immediate early events following cell-surface binding and viral cellular process signalling that, as in the case of ACE2, LAPase is also involved in Angiotensin II degradation and early events of the HIV viral entry pathway[17-19,57-58].
Although, at present, more robust evidence is needed to further assess the potential role of LAPase in SARS-CoV-2 infectivity, there is circumstantial evidence to indicate that individuals suffering from obesity and some types of cancer may have benefited from the inclusion of selective gut microbiota nNE biogenic food adjuvants as part of their controlled diets, some of which may reduce inflammation by the inhibition of LAPase activity.
Out of eight gut microbiota nNE biogenic clones, we are currently in the process of seeking the industrial manufacturing of selective nNE biogenic clones that may function as beneficial adjuvants to enhance the effectiveness of continuing vaccination strategies and, possibly, address some of the early and enduring ailments observed in patients suffering from Post-COVID-19 Syndrome triggered by SARS-CoV-2 induced hyperinflammation.
In earlier reports, we described the critical steroid interdependency of LAPase activity and its role in the early detection of breast cancer and the susceptibility of women towards HIV infection[17-19,59-61]. These studies showed that LAPase activity levels determined in sera obtained from HIV patients were up to three orders of magnitude higher when compared to control uninfected subjects, with women being more susceptible to HIV infectivity[17-19]. In addition, cellular and extracellular LAPase activities were significantly higher in infected cells when compared to uninfected human lymphocytic HUT78 cells whereby treatment with Bestatin [(2S, 3R)-3-amino-2-hydroxy-4-phenyl-butanoyl-l-leucine] hindered HIV infection[17-19].
The prospect of further assessing the anticipated role of LAPase in the immediate viral entry of SARS-CoV-2 has several important clinical ramifications. Apart from eliciting the design of new therapeutic compounds, it can provide a better understanding of some of the lingering symptoms observed in patients suffering Post COVID-19 Syndrome primarily affecting women[49,62,63].
Due to the lack of a refined assessment clinical tool, it is a current practice for individuals who test positive for COVID-19 to be directed to return home without further clinical assessment. Most COVID-19 positive patients only return to clinical care once they experience an advanced symptomatology. We propose to perform routine LAPase activity in sera of COVID-19 patients commencing on the day of a confirmed positive result and during the first seven days post-infection regardless of health status. This information could provide a quantitative predictive value concerning COVID-19 positive patients that might require early clinical attention due to a potential poor prognosis for a recovery without requiring hospitalisation as well as predicting the possible onset of lingering hyperinflammation leading to Post COVID-19 syndrome.
Recent studies focused on the potential role of neutrophils in SARS-CoV-2-induced hyperinflammation also described the potential role of six other SARS-CoV-2 putative receptors in addition to ACE2[64]. These include two peptidases (DPP4 and ANPEP) as well as 4 pathogen binding proteins (CD209, CLEC4G, CLEC4M, and CEACAM1)[64].
Interestingly, using bronchoalveolar lavage fluid cells from COVID-19 patients, extensive gene mapping and expression by means of RNA-seq data set analysis revealed upregulation of a variety of neutrophil inflammatory genes including LAP3 gene encoding Cytosol Aminopeptidase[64]. These findings, combined with the implication of LAPase in HIV viral entry and infectivity, further support the rationale of measuring circulating LAPase activity in sera of COVID-19 patients commencing on the day of a confirmed positive result and during the first seven days post-infection regardless of health status in order to rapidly and quantitatively predict disease severity and proceed with currently available adjuvant therapies prior to the onset of more severe symptoms.
Apart from monitoring LAPase activity in sera of COVID-19 patients immediately after infection, it is of great interest to determine LAPase levels in patients suffering from Post-COVID-19 Syndrome and to clinically evaluate the use of Bestatin [(2S, 3R)-3-amino-2-hydroxy-4-phenyl-butanoyl-l-leucine] in reducing lingering symptoms.
Clinical Significance of Emerging Variants and Vaccination Strategies against COVID-19
The considerable amount of SARS-CoV-2 global infections, reaching close to 200 million reported cases, together with the evidence of rapid viral adaptive fixation of favourable Single Nucleotide Variations (SNVs) most likely dependent upon clonal interference and convergent evolution processes have led to thousands of newly reported genomic variants (see Table 3 and Figures 1A and 1B), ensuring the long-lasting prevalence of SARS-CoV-2 in the months and, most likely, years to come.
As described in previous sections, the rapid ability of SARS-CoV-2 to continue achieving positive adaptive evolution through its capacity to co-transmit new favourable variations on the same haplotypes during infection is of particular concern.
This level of successful and continuous viral adaptive evolution could eventually lead to the emergence of SARS-CoV-2 super-variants with more far-reaching health consequences than those currently identified as variants of interest and variants of concern (see previous sections and Table 1), potentially creating variants that reach the status of SARS-CoV-2 Variants of High Consequence.
In addition, the role that we propose for selective aminopeptidases belonging to the M1 family, such as Leucine Aminopeptidase (LAPase), capable of playing a role as an immediate viral entry accessory receptor (IVEAR) independent of the ACE / Neuropilin-1-dependent pathway, signals the equal potential for the emergence of a new Sarbecovirus lineage with the capacity to become a pathogen of significance to global health.
It is, therefore, of utmost relevance not only to continue performing genomic analysis of emerging SARS-CoV-2 infection clusters but also to institute global, robust and centralised detection and reporting of new SARS-CoV-2 SNVs.
The severity of the unremitting COVID-19 pandemic outbreak has unleashed the most unifying and prolific output by scientists around the world, reflected in the swift development of rapid and accurate screening tools, adjuvant biotherapeutics and vaccines.
In a series of impressive and concerted efforts, from the design and development of an array of vaccines to the swift regulatory approval and logistical support needed to mount effective global vaccination campaigns, as shown in Table 5 (A-D), at least 11 different vaccines have already reached the arms of a significant number of people in various countries.
Moreover, this first round of vaccines has used an array of different biological technologies, such as mRNA (Table 5A), recombinant replication deficient adenoviral vectors (Table 5B), protein nano-engineering / chemically-derived synthetic peptide vaccines (Table 5C) and viral expansion and inactivation (Table 5D).
The level of safety and efficacy, including protection against current variants of concern, outlined in Table 1 is asymmetrical, and it is still in the process of being more accurately assessed as the population of vaccinated individual increases and the post-vaccination surveillance and reporting of non-serious and serious adverse events continues to be made public (refer to Table 5 A-D for details).
5A
| Manufacturer | Name | Technology | Doses | Efficacy |
| Pfizer/BioNTech | BNT162b2 | mRNA | 2 |
Documented Infections[65] Female 14 to 20 days after first dose 50%. 7 days after second dose 93%. Male 14 to 20 days after first dose 41%. 7 days after second dose 91%. Symptomatic Illness[65] Female 14 to 20 days after first dose 60%. 7 days after second dose 96%. Male 14 to 20 days after first dose 52%. 7 days after second dose 88%. Protection Against Variants B.1.1.7, B.1.351 and B.1.1.28/B.1.1.248[66] |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
| 13.6 | 4.70 | 0.35 | 4349 |
5A
| Manufacturer | Name | Technology | Doses | Efficacy |
| Moderna/NIH | mRNA-1273 | mRNA | 2 |
94.1%[67] Protection Against Variants B.1.1.7, B.1.351 Protection against P.1, B.1.427 / B.1.429 variants remains to be determined[68]. |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
| 36.94 | 3.44 | 0.09 | 2418 |
5B
| Manufacturer | Name | Technology | Doses | Efficacy |
| Oxford/AstraZeneca |
AZD1222 ChAdOx1 nCoV-19 |
Recombinant, replication-deficient chimpanzee adenovirus vector | 2 |
Interim Report 3 Continents
Symptomatic Illness[66] 64.1% after first standard dose 70.4% after second standard dose Protection Against Variants 60% effective against B.1.617.2 two weeks after the second dose 66% against B.1.1.7 variant and 33% effective against symptomatic disease from B.1.617.2 three weeks after the first dose[70] No apparent protection against mild-to-moderate Covid-19 due to the B.1.351 variant[71] |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
| 28.57 | 12.44 | 0.44 | 1065 |
5B
| Manufacturer | Name | Technology | Doses | Efficacy |
| Gamaleya RI | Gam-COVID-Vac | Two replication-deficient (rAd26 and rAd5) human adenoviral vectors | 2 |
Interim Analysis Randomised Controlled Phase III Trial (Russia)[72] Undesirable induction of immune response against viral vector minimised by the use of two vectors: First dose (rAd26) Second dose (rAd5) both containing full length SARS-CoV-2 Spike glycoprotein Efficacy 21 Days after First Dose (Day of Second Dose) was 91.6%. Protection Against Variants Unknown. Data discrepancies concerning the Interim Analysis Phase III Trial have been reported and addressed[73]. |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
|
Data from Canada not available. Based upon interim analysis of controlled Phase III Trial (Russia), 94% of adverse events were reported as being of Grade 1 (Scale 1-5; 1= less), being of lesser concern[72]. |
Data from Canada not available. Based upon interim analysis of controlled Phase III Trial (Russia), 0.3% were ranked as serious with death events reported as unrelated events[72]. |
Data from Canada not available. | Data from Canada not available. |
5B
| Manufacturer | Name | Technology | Doses | Efficacy |
| Johnson & Johnson | JNJ-78436735 | Replication-incompetent human adenoviral vector (Ad26.COV2.S) | 1 |
Interim Analysis of Phase III Clinical Data Eight Countries: Argentina, Brazil, Chile, Colombia, Mexico, Peru, South Africa, and the United States. Total 44,325 adult volunteers[74]. Efficacy 28 Days Post-Vaccination was 66% at preventing moderate to severe COVID-19 and 85% protective against most severe COVID-19 symptoms[74]. Level of protection varied regionally: 72% in the United States, 66% in Latin American countries and 57% in South Africa[74]. Protection Against Variants Protection against B.1.351 variant estimated at 57%[74]. |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
|
Data from Canada not available. Based upon interim analysis of Phase III Clinical Data from eight countries, only mild side effects were reported[74]. |
Data from Canada not available. Sixteen deaths in placebo group compared to three deaths in the vaccine group[74]. |
Data from Canada not available. | Data from Canada not available. |
5B
| Manufacturer | Name | Technology | Doses | Efficacy |
| CanSinoBio | AD5-nCOV | Replication-defective Ad5 viral vectors expressing the full-length spike gene (Wuhan-Hu-1) | 1 |
Interim Analysis of Phase III Clinical Data. Six Countries: Argentina, Chile, Mexico, Pakistan Russia and Saudi Arabia. Total 40,000 participants. Efficacy in Preventing Symptomatic Disease 65.7% Stopping Severe Disease 90.98%[79] Protection Against Variants According to the Chinese Center for Disease Control and Prevention, AD5-nCOV vaccine provides protection against current variants, being less effective against the B.1.617.2 / Delta variant. |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
|
Data from Canada not available. As reported in a randomised, double-blind, placebo-controlled Phase II trial: 9% severe adverse reactions found in participants given a 1 x 1011 viral particles dose and 1% of participants in the 5 x 1010 group[80]. |
Data from Canada not available. No severe reactions reported in a randomised, double-blind, placebo-controlled Phase II trial[80]. |
Data from Canada not available. | Data from Canada not available. |
5C
| Manufacturer | Name | Technology | Doses | Efficacy |
| Novavax | NVX-CoV2373 | Protein nano-engineering of a SARS-CoV-2 subunit (NVX-CoV2373) from the full-length spike (S) protein forming 27.2-nm thermostable nanoparticles | 2 |
Interim Analysis of Phase III Clinical Data (UK), 15,000 participants aged 18-84 (27% over 65), 95.6% against original SARS-CoV-2 variant[75]. Protection Against Variants B.1.1.7 (85.6%) and B.1.351 (60%)[75] |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
|
Data from Canada not available. Interim Analysis of Phase III Clinical Data: United Kingdom, Mexico and the United States have not reported any significant numbers of severe, serious and medically attended events. |
Data from Canada not available. In January, 2021, Novavax formally commenced registration of NVX-CoV2373 with the UK Medicines and Healthcare Products Regulatory Agency. Low numbers of serious adverse events appear to have been reported with equivalent proportions shared between vaccine and placebo groups[75]. |
Data from Canada not available. | Data from Canada not available. |
5C
| Manufacturer | Name | Technology | Doses | Efficacy |
| VECTOR Centre of Virology | EpiVacCorona | Chemical synthesis of three small Spike protein peptides conjugated to a carrier polypeptide and absorbed into Aluminium hydroxide | 2 | Pending completion and publication of ongoing Phase III clinical trial involving more than 3,000 participants |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
| Data from Canada not available. | Data from Canada not available. | Data from Canada not available. | Data from Canada not available. |
5D
| Manufacturer | Name | Technology | Doses | Efficacy |
| Bharat Biotech | Covaxin | Inactivated SARS-CoV-2 containing adjuvant(Aluminium hydroxide) | 2 |
Interim Analysis and Report of Phase III Clinical Data The Indian Council of Medical Research (ICMR) recently reported an efficacy of 81% in preventing COVID-19. Data obtained from 21 sites involving 25,800 subjects[76]. Protection Against Variants hCoV-19/India/20203522 (UK-variant) and hCoV27 19/India/2020Q111 (heterologous strain)[77] |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
|
Data from Canada not available. Safety and immunogenicity double-blind, randomised Phase I trial: mild (69%), moderate (31%)[78]. |
Data from Canada not available. No serious adverse reactions reported as of to date[78]. |
Data from Canada not available. | Data from Canada not available. |
5D
| Manufacturer | Name | Technology | Doses | Efficacy |
| Sinovac | CoronaVac | Inactivated vaccine candidate against COVID-19, produced through SARS-CoV-2 (CN02 strain) inoculation and viral expansion using African green monkey kidney cells (Vero cells), harvested virus inactivated with β-propiolactone, concentrated, purified and absorbed into aluminium hydroxide-based adjuvant | 2 |
Pending analysis of approval of emergency use of CoronaVac in China and analysis of three ongoing Phase III clinical trials: Brazil (NCT04456595) Indonesia (NCT04508075) Turkey (NCT04582344)[81] Protection Against Variants 50% efficacy against P.1 Brazilian, Gamma variant[84] |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
|
Data from Canada not available. Analysis of a randomised, double-blind, placebo-controlled Phase I / II clinical trial in adults 18-59 reported mild Grade 1 (Scale 1-5; 1 = less) adverse reactions: 19% in 3 μg group; 19% in 6 μg group; 18% in placebo group (0 and 28 days)[81] |
Data from Canada not available. No vaccine-related serious adverse events were reported within 28 days of second dose[81]. |
Data from Canada not available. | Data from Canada not available. |
5D
| Manufacturer | Name | Technology | Doses | Efficacy |
| Sinopharm | BBIBP-CorV | Inactivated vaccine using three SARS-CoV-2 strains obtained from bronchoalveolar lavage samples or throat swabs of three hospitalised patients: 19nCoV-CDC-Tan-HB02 (HB02), 19nCoV-CDC-Tan-Strain03 (CQ01) and 19nCoV-CDC-Tan-Strain 04 (QD01). Propagation of HB02 on WHO Certified Vero Cells was performed for industrial manufacturing. Inactivated purified virus is absorbed into aluminium hydroxide-based adjuvant. | 2 |
Vaccine efficacy in multi-country Phase II Trial[82] Overall 78.1% Hospitalisation 78.7% Sex Male 78.4% Female 75.6% Age Group 18-59 years 78.1% Comorbidities Diabetes 63.7% Obesity 80.7% Protection Against Variants Pseudo-virus neutralisation assays seem to provide neutralisation against B.1.351. Interim efficacy against variants of concern not assessed during Phase III Clinical Trials[82]. |
| Total Non-Serious Adverse Event Reports Rate* | Total Serious Adverse Event Reports Rate* | Total Serious / Non-Serious Rate Ratio* | Total Number of Reports | |
|
Data from Canada not available. According to a randomised, double-blind, placebo-controlled Phase I / II trial[83], adverse reactions were as follows: Any 29% Grade 1 21% Grade 2 8% |
Data from Canada not available. To be reported. |
Data from Canada not available. | Data from Canada not available. |
Table 5 Characteristics of Approved and Clinically Advanced Vaccines
Table 5 (A-D) summarises relevant characteristics and clinical data available related to 11 approved vaccines currently deployed in vaccination campaigns in various countries. These encompass RNA vaccines (Table 5A), DNA adenoviral vector vaccines (Table 5B), protein nano-engineering / chemically-derived synthetic peptide vaccines (Table 5C) and viral expansion and inactivation vaccines (Table 5D). Some vaccines have only been granted approval in a limited number of countries on the basis of emergency use authorisation granted by their corresponding health regulatory government departments. Although protection against current predominant variants such as B.1.1.7, B.1.351 and B.1.1.28/B.1.1.248 has been documented, not all 11 vaccines confer the same level of protection against these and emerging SARS-CoV-2 variants. Three main vaccines are currently being used in Canada in ongoing, extended vaccination campaigns including individuals 12 years of age and older. These are described in Table 5A (RNA vaccines from Pfizer/BioNTech and Moderna/NIH) and Table 5B (DNA adenoviral vector vaccine from Oxford/AstraZeneca), including detailed cumulative safety and efficacy data collected.
*per 100X103 doses administered within Canada from January 1, 2021, to June 18, 2021.
The relentless, asymmetric and sustained SARS-CoV-2 global infectivity soon to reach 200 million infected individuals and the proven efficacy of the first round of vaccines described in Table 5 (A-D) to decrease infectivity and mortality rates, have sparked a swift second wave of hundreds of new vaccine types, including 105 in clinical development and 184 in pre-clinical development.
In addition, several concerted efforts have been made in a global effort to secure greater vaccination rates and percentages of people being vaccinated, currently sitting at 13% of the population in the world being fully vaccinated.
Some of the most relevant global efforts to secure a diverse and robust source of funding for vaccine development, equal access and distribution of successful vaccines include:
The scope and magnitude of these concerted international efforts is a reflection of the immediate and long-term impact of emerging SARS-CoV-2 variants and of the increasing importance of the expected emergence of a novel Sarbecovirus lineage to Global Human and Animal Health.
Promptly securing an array of SARS-CoV-2 vaccines will not only increase our current ability to increase global vaccination rates per country but will enhance the current capacity and readiness to mitigate the ability of SARS-CoV-2 genomic adaptive evolution through clonal interference and molecular convergent evolution leading to more contagious and aggressive variants.
Furthermore, although current vaccines seem to continue to provide different levels of protection against variants of concern, fully vaccinated individuals continue to be infected with SARS-CoV-2[85]. Therefore, it is conceivable that even a vaccinated population could provide an opportunity for SARS-CoV-2 to mutate, as well as enabling asymptomatic transmission.
Viral load measurements from infected individuals with current predominant variants described in Table 1 after partial and full vaccination with the various vaccines available remain to be determined. However, in a population of vaccinated individuals 16-89 years old, it appears that vaccination with the BNT162b2 mRNA vaccine results in reduced viral load infectivity 12-37 days after the first vaccination[86].
In this regards, although the minimum infectious SARS-CoV-2 viral particles eliciting a full blown infection remains to be determined, it is highly anticipated that this number will depend upon the type of SARS-CoV-2 predominant variant at any given time. Current available data suggest that, on average, infectivity and transmission can take place with less than 100 viral particles[87].
Although the upcoming results of an ongoing live SARS-CoV-2 infectivity challenge study performed in humans are expected to shed light on the minimum effective viral loads required to elicit infection[88], it is already known that infectious viral particles can be detected in faeces from infected patients even after negative polymerase chain reaction (PCR) testing results from bronchoalveolar lavage samples or throat swabs[89].
This observation has led to the articulation of a Global SARS-CoV-2 Wastewater Monitoring Effort encompassing 263 universities in 55 countries, including Canada.
This sentinel approach might not only provide accurate information of anticipated regional infectivity clusters but also insight into the potential isolation and characterisation of SARS-CoV-2 variants of interest. Data from this effort might be able to offer a direct input into ongoing vaccine development efforts to better address the continuous emergence of variants of concern.
As some countries continue to reduce public health measures based upon their current full vaccination rates and mounting social pressures to resume a higher level of social and economic normalcy, based upon the sustained and significant global level of SARS-CoV-2 infections together with the fact that only 13% of the global population has been vaccinated as of July, 2021, as in the summer of 2020, the summer of 2021 will most likely provide the optimal environment for the emergence of yet another cluster of SARS-CoV-2 variants of concern.
Based upon the anticipated role of Leucine Aminopeptidase (LAPase) in the immediate viral entry of SARS-CoV-2 independent of the S1 CendR motif required for SARS-CoV-2 binding and internalisation through the Neuropilin-1-dependent pathway, it is of great interest to consider the relevance of amino-terminal domain mutations (NTDs) presently occurring in the SARS-CoV-2 Spike (S) protein in the design of a new wave of SARS-CoV-2 vaccines.
Similar observations have been recently reported, noting that not only there has been an increased frequency of the NTD mutation L18F in both B.1.1.7 and B.1.351 but also that NTD mutations seem to also confer reduced sensitivity to neutralising antibodies[90].
In a previous report focused on a dynamic multifactorial model of the origins of SARS-CoV-2, we emphasized the relevance of SARS-CoV-2 intra- and inter-species transmission between humans and domesticated animals such as cats and ferrets, leading to subsequent intra-species infections and transmission cats-to-cats and ferrets-to-ferrets, with the potential inter-species SARS-CoV-2 transmission from these infected domesticated animals back to humans[4].
It is during the relaxation of health measures in the summer of 2020 and due to overcrowding conditions prevailing on mink farms in Europe and North America that the most relevant inter-species transmission of SARS-CoV-2 took place, affecting people involved in the farming, culling and pelting of mink, resulting in the deplorable mass culling of over 17 million mink animals[27,41-43].
In addition to this tragic loss of mink animals, the emergence of a new variant, identified as the ΔFVI spike/Cluster 5 variant (see Table 1), led to the rapid transmission of the mink-related mutation Y453F in the viral spike found in humans as well as being one of the 170 mutations identified after performing whole genome sequencing of mink SARS-CoV-2 samples obtained from 40 mink farms[91]. The sharing of the mutation Y453F in the viral spike in SARS-CoV-2 samples obtained from mink and humans is of relevance in two fronts.
First, it strengthens the concept of inter-species transmission of mutations of relevance, most likely through positive adaptive episodes by clonal interference and molecular convergent evolution rather than solely by the emergence of host-driven viral evolution through intra-species, post-infection mutations.
Second, it reinforces the importance of routine screening and genomic surveillance that includes domesticated animals, with the purpose of better predicting the emergence of SARS-CoV-2 variants of concern as well as to better refine the development of a new generation of vaccines.
In the specific case of the use of animals such as mink for the sole purpose of manufacturing luxurious garments, it is suggested that this practice be replaced by the use of more durable luxurious man-made textiles that exclude the use of animal fur.
Although some European countries appear to have embraced the cessation of mink farming, the emergence of zoonotic diseases of the magnitude of SARS-CoV-2 calls for a global and concerted effort to decrease, on the one hand, the production of animal-based foods in over-crowded industrial conditions and, on the other hand, the current level of exploitation of marine species for human consumption. Recent estimates indicate that 77 billion land animals and 1.2 trillion aquatic animals are killed yearly for human consumption[92,93].
Apart from the continuous refinement of the safety and efficacy of current and emerging vaccines to address the continuous rise of SARS-CoV-2 variants of concern, based upon the preliminary results of small clinical trials, several countries, including Canada, have endorsed the mixing of two different vaccines such as an initial DNA adenoviral vector vaccination with the Oxford-AstraZeneca vaccine followed by Pfizer-BioNTech mRNA lipid nanoparticles containing Polyethylene Glycol (PEG)[94].
In Canada, this approach resolved COVID-19 vaccine hesitancy due to higher Total Serious / Non-Serious Rate Ratios and Total Serious Adverse Event Reports Rates by comparison to mRNA vaccines as well as a perceived lesser degree of protection against variants of concern using the Oxford-AstraZeneca vaccine alone (see Table 5).
An additional level of COVID-19 vaccine hesitancy relates to the vaccination of younger individuals encompassing the age group between 12 and 17 years of age[95]. Although children seem to express lower levels of ACE2, the putative receptor of SARS-CoV-2, and, hence, seem to be less exposed to SARS-CoV-2 infections, such decreased expression seems to be asymmetrical, with differences being found between nasal and pulmonary epithelium[95].
While decreased ACE2 expression in the nasal epithelium may result in less susceptibility to SARS-CoV-2 infections, a decreased expression in the pulmonary epithelium may actually result in more severe disease because soluble ACE2 could lead to clearance of SARS-CoV-2 viral particles from pulmonary epithelium cells[95].
Although a new study, entitled Human Epidemiology and Response to SARS-CoV-2 (HEROS), will help to better understand the susceptibility towards SARS-CoV-2 infection in relation to age and levels of ACE2 expression, to date, children, adolescents and young adults (0-24) accounted for almost 3 million COVID-19 cases in the USA, alone, from March to December, 2020[96], prompting vaccination roll-outs in individuals 12 years and older.
Out of all 11 vaccines summarised in Table 5 and currently being used in global vaccination campaigns, Canada vaccination roll-outs have used two mRNA vaccines (Pfizer/BioNTech BNT162b2 and Moderna/NIH mRNA-1273) and one DNA Adenoviral vector vaccine (Oxford/AstraZeneca AZD1222 ChAdOx1 nCoV-19).
Although the safety of all approved vaccines in Canada continues to track according to expected low levels of overall non-serious and serious event reports (see Table 5 A-B), there appear to be important distinctions. These comprise:
Taking into account vaccination hesitancy across Canada during early vaccination roll-outs, certain provinces, such as Ontario, opted to endorse the mixing of two different vaccines so that people who had received a first dose of the Oxford-AstraZeneca DNA adenoviral vector vaccine received Pfizer-BioNTech or Moderna/NIH mRNA vaccines as their second vaccination dose. This vaccination strategy seems to have positively impacted vaccination rates, leading to a total of 71% of the population having received one dose (16%) or two (55%) as of July 24, 2021.
As global post-vaccination surveillance continues to be actively pursued by the international scientific research community, the incidences of undesirable rare events have been recently characterised. One such event encompasses the onset of autoimmune-like heparin-induced thrombocytopenia apparently mediated by platelet-activating antibodies against Platelet Factor 4 (PF4) after Oxford/AstraZeneca vaccination[97].
With regard to mRNA vaccines, during the processing of mRNA encapsulated in nanoparticles or liposomes, prior to reaching the ribosomal complexes to initiate translation, mRNA molecules could be bound to endosome or cytosolic Pattern Recognition Receptors (PRRs) such as Toll-like receptor (TLR) 3, TLR7 and TLR8 within endosomes or to retinoic acid inducible gene-I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) in the cytosol[98,99].
These exquisite and concerted intracellular receptor signalling pathways involving mRNA molecules trigger an array of pro-inflammatory cascades, including the assembly of inflammasomes, playing a crucial role in the initiation of innate immune responses normally initiated by the detection of potentially harmful pathogens[98,99]. In the case of mRNA vaccines, it has been postulated that these intracellular signalling pathways could lead to an increased risk of immune-mediated ailments including autoimmune diseases, resulting in the recommendation that young female patients already affected or predisposed to immunological disorders carefully evaluate the benefits and risks associated with mRNA COVID-19 vaccination[98,99].
In the specific case of individuals suffering from rheumatoid arthritis, patients receiving various combinations of immunosuppressive treatments such as methotrexate alone or in combination with abatacept, Janus kinase inhibitors, prednisone or any continuous therapy with conventional synthetic, biological or targeted synthetic disease-modifying anti-rheumatic drugs (DMARDs), antibody titres against SARS-CoV-2 spike protein 1 and for SARS-CoV-2 nucleoprotein were significantly lower after first and second vaccinations with mRNA-1273 vaccine (Moderna/NIH) or BNT162b2 vaccine (Pfizer/BioNTech) by comparison with controls[100]. Based upon these observations, it has been proposed that rheumatoid arthritis patients under continuous immunosuppressive treatments timely receive two vaccinations within an interval of 3-6 weeks[100].
As we continue to develop global vaccination campaigns to address the current unyielding COVID-19 pandemic outbreak, the tailoring of improvements to vaccines currently being used in vaccination roll-outs to better address the continuous emergence of SARS-CoV-2 variants should continue to take into account the potential onset of undesirable side effects, in particular those related to thrombotic thrombocytopenia and autoimmune related disorders.
The impressive pipeline of new vaccines currently under development will increase our ability to rapidly respond to adjustments in vaccination strategies in order to better address specific regional infection clusters in accordance with the particular health status of targeted populations.
Conclusions
As stated in some of our recent publications underscoring critical events related to the immediate viral entry of SARS-CoV-2 and the possible multifactorial origins of the S (Serine) and L (Leucine) variant types of SARS-CoV-2, based upon available clinical data at that time, we estimated that December 8, 2019, signalled the beginning of one of the most acute human global health challenges in decades with the earliest publicly available clinical reports of patients suffering from pneumonia of unknown origin[1-6].
Since then, two compelling and eloquent scientific contributions have significantly improved our current understanding of pivotal events leading to the successful adaptive evolution of SARS-CoV-2 and its sustained capacity to elicit both inter-species (animals-to-humans) and intra-species (humans-to-humans, animals-to-animals) infectivity and transmission of current and ancestral SARS-CoV-2 variants.
The first relates to the extraordinarily high allelic association (R2 = 0.987) shared between two SARS-CoV-2 Single Nucleotide Variations (SNVs) identified as C8782T (Serine) and C28144C (Serine) (21).
Apart from being detected in perhaps one of the first SARS-CoV-2 variants (MT291826) in addition to that originally isolated on December 26, 2019, and described under the name of WH-Human 1 coronavirus[24], based upon their high coefficient of allelic association, it is highly likely that SNVs C8782T (Serine) and C28144C (Serine) were co-transmitted during initial and subsequent infection episodes with concurrent or sequential incidences leading to in vivo asymmetrical mutation patterns of ancestral forms of SARS-CoV-2 that were most likely close to the S (Serine) ancestral Type[4,21,22].
This is of great relevance to better understand the early adaptive evolution of SARS-CoV-2. Both SNVs (C8782T and C28144C) were actually used to designate the S (Serine) and L (Leucine) Types of SARS-CoV-2, whereby the S (Serine) Type, classified within the viral strain Type II, is not only closer to bat SARSr-CoV RaTG13 but is, most likely, related to the original strain of SARS-CoV-2 infecting humans following concurrent asymmetrical patterns of transmission[4,21,22].
It is highly likely that pinpointing the accurate origins of SARS-CoV-2 will continue to remain challenging. This is due in part to the fairly rapid closure and sanitisation of all possible affected areas within the province of Hubei where the first clinical cases of COVID-19 were documented.
Nonetheless, based upon some of the main characteristics of the sabercovirus phylogenetic tree (partially summarised in Figures 2A and 2B), we proposed two sets of comparative studies to evaluate the in vitro replication and cytopathicity in Vero E6 and Calu-3 2B4 cells at low multiplicity of infection (0.01 plaque-forming units [PFU] per cell) as well as a second set of similar in vivo studies using the hamster model of SARS-CoV-2 pathogenesis[51] using the following Sarbecoviruses:
In addition, due to the potential early co-transmission of SNVs C8782T (Serine) and C28144C (Serine) found in the otherwise ancestral S (Serine) Type, it is of equal importance to evaluate the replication and cytopathicity of single mutations involving SNV C8782T alone, SNV C28144C alone and those in which both potentially co-transmitted SNVs are deleted.
The results of these study sets could provide a more accurate picture regarding the origins of SARS-CoV-2.
The second most relevant scientific contribution pertains to the generation of a polybasic Furin mutant lacking the Furin (S-Pro-Arg-Arg-Ala-R/PRRA) cleavage site motif (ΔPRRA) using a SARS-CoV-2 reverse genetic system[14]. This study clearly demonstrated the ability of ΔPRRA to elicit attenuated infections both in human cells, including Calu-3 human lung cancer cell lines, and in animal models[14].
The ability of ΔPRRA to elicit attenuated infections both in vitro and in vivo is important for two reasons. First, it provides evidence that the absence of the polybasic Furin (S-Pro-Arg-Arg-Ala-R/PRRA) cleavage site motif in bat SARSr-CoV RaTG13 and other coronaviruses of the same clade does not hamper the possible direct primordial infection of an ancestral SARSr-CoVRaTG13 from bats to humans followed by human adaptive evolution through human-to-human super-spreader-mediated transmission leading to the emergence of the ancestral S (Serine)/Type II variants of SARS-CoV-2[4]. Second, the deletion of the polybasic Furin (S-Pro-Arg-Arg-Ala-R/PRRA) cleavage site hinders the sequential release of the S1 CendR motif required for SARS-CoV-2 binding and internalisation through the Neuropilin-1-dependent pathway due to the removal of the Furin cleavage from the S1/S2 junction of SARS-CoV-2 RBD[14,52]. Since ΔPRRA elicits attenuated infections both in vitro and in vivo, we have proposed the existence of Neuropilin-1 independent immediate viral entry accessory receptors (IVEARs). Specifically, we anticipate the role of selective aminopeptidases belonging to the M1 family, such as Leucine Aminopeptidase (LAPase), to be that of immediate viral entry accessory receptor(s) (IVEARs) independent of the ACE2/Neuropilin-1-dependent pathway.
Apart from its involvement in Angiotensin II degradation, our proposal is founded upon ample evidence that Leucine Aminopetidase (LAPase) can also potentiate HIV infectivity during immediate early events following cell-surface binding and cellular-mediated viral processing[17-19,57,58].
Whilst more robust clinical and experimental evidence is required to further substantiate the role of LAPase in SARS-CoV-2 infectivity, there is circumstantial evidence to indicate that individuals suffering from obesity and some cancer types may have benefited from the inclusion of selective gut microbiota nNE biogenic food adjuvants as part of their controlled diets, some of which may reduce inflammation by inhibition of LAPase activity.
From eight gut microbiota nNE biogenic clones, we are currently advancing the industrial manufacturing of selective nNE biogenic clones, some of which may act as beneficial adjuvants to enhance the effectiveness of continuing vaccination strategies and, possibly, address some of the early and enduring ailments observed in patients suffering from Post-COVID-19 Syndrome triggered, amongst other things, by SARS-CoV-2/neutrophil-induced hyperinflammation.
Due to the early events potentially leading to enhanced LAPase activity following SARS-CoV-2 infectivity, we propose to measure serum LAPase activity in all patients who have tested positive against SARS-CoV-2 as an important clinical tool for the early prediction of patients who may develop severe hyperinflammation and, hence, commence early clinical care as soon as possible.
To date, it is a current practice that persons who test positive for COVID-19 are left to return home without further clinical assessment. Most COVID-19 positive patients only return to clinical care once they experience advanced symptoms. Hence, we propose to perform routine measurements of LAPase activity in sera of COVID-19 patients, commencing on the day of a confirmed positive result and during the first seven days post-infection, regardless of health status.
This information may offer a quantitative predictive value concerning COVID-19 positive patients who might require early clinical interventions due to a potentially poor prognosis for recovery, without requiring hospitalisation as well as flagging the possible onset of lingering hyperinflammation characteristic of Post COVID-19 Syndrome.
Routinely measuring serum LAPase activity in patients suffering from Post-COVID-19 Syndrome could also represent an extremely valuable tool for better understanding the evolution of hyperinflammation processes as part of the lingering symptoms observed in patients suffering from this syndrome. This could be particularly beneficial for women who have been reported to be the most affected by this condition due to the reported oestrogen interdependency of LAPase activity and enhanced viral entry of other viruses such as HIV[17-19,49,57,58,62,63].
The potential accessory role of LAPase to the immediate viral entry and infectivity of SARS-CoV-2 could assist in the better design of adjuvant therapies including the use of nNE biogenic clones as well as the use of known inhibitors of LAPase such as Bestatin.
Reaching globally close to 200 million people infected with SARS-CoV-2, with merely 14% of individuals being fully vaccinated against COVID-19 around the world, the efficient and fast-paced viral adaptive fixation of favourable Single Nucleotide Variations (SNVs), most likely driven by clonal interference and convergent evolution mechanisms, has already resulted in thousands of newly reported genomic variants (see Table 3 and Figures 1A and 1B), securing a long-lasting prevalence of SARS-CoV-2 for the months and, most likely, years to come.
In addition, the remarkable ability of SARS-CoV-2 to continue securing positive adaptive evolution through its capacity to co-transmit new favourable variations on the same haplotypes during infection is troublesome at two levels. Such a continuously successful viral adaptive evolution could eventually lead to the emergence of SARS-CoV-2 super-variants with far reaching health consequences even more significant than those currently identified as variants of interest and variants of concern, potentially attaining the status of SARS-CoV-2 Variants of High Consequence. Furthermore, the anticipated role of selective aminopeptidases belonging to the M1 family, such as Leucine Aminopeptidase (LAPase) capable of playing a role as an immediate viral entry accessory receptor (IVEAR) independent of the ACE2/Neuropilin-1-dependent pathway, signals, equally, the potential emergence of a new Sarbecovirus lineage with the capacity to become a pathogen of global health significance.
The emergence of ΔFVI spike/Cluster 5 SARS-CoV-2 variant during the summer of 2020 provided a clear example of the relevance of inter-species viral transmissions. As we had previously underlined in our dynamic multifactorial model on the origins of the S and L Types of SARS-CoV-2, inter-species transmissions provide yet another avenue for effective adaptive evolution, enabling the emergence of potential SARS-CoV-2 variants of concern, raising awareness of unnecessary farming practices such as the overcrowded exploitation of mink for their pelts for luxury garments.
In June, 2020, during the relaxation of preventative health measures in Europe, the appearance of ΔFVI spike/Cluster 5 SARS-CoV-2 led to the unprecedented culling of more than 17 million mink animals, the destruction of raw pelts, the quarantine of mink workers and the closure of mink farms and related facilities[27,41-43]. The rapid inter-species transmission of human infections such as SARS-CoV-2 to highly confined animals such as mink underscores the pressing need to halt the use of raw pelts obtained from live animals instead of man-made, durable and highly performant materials currently available for tailoring every day and luxury garments.
The onset of the ΔFVI spike/Cluster 5 SARS-CoV-2 variant also confirms our previous concern that anthropogenic activities could lead to both inter-species (animals-to-humans-to-animals) and intra-species (humans-to-humans, animals-to-animals) infectivity and transmission of current and ancestral SARS-CoV-2 variants[4]. Apart from mink, it is known that SARS-CoV-2 inter-species infections between humans and domesticated animals with subsequent intra-species SARS-CoV-2 transmission and infectivity can involve cats and ferrets, as well[4].
Although some European countries seem to have embraced the cessation of mink farming, the emergence of zoonotic diseases of the magnitude of SARS-CoV-2 calls for a global and concerted effort to decrease, on one the hand, the production of animal-based foods in overcrowded industrial conditions and, on the other hand, the current level of exploitation of marine species for human consumption. Recent estimates indicate that 77 billion land animals and 1.2 trillion aquatic animals are killed yearly for human consumption[92,93]. Not only is this practice clearly unsustainable, it will continue to lead to more frequent zoonotic diseases whilst also accelerating the looming extinction of one million plant and animal species within only a few decades[103].
By the end of 2019, under the imminent threat of SARS-CoV-2 to human health, the global scientific community sparked a relentless and remarkable set of unique, innovative achievements, enabling a swift response to identify, characterise, diagnose and treat SARS-CoV-2. Clinical research organisations and regulatory authorities, as well as operational and logistical entities around the world, truly enabled the successful deployment of the first wave of vaccine roll-outs, from the initial design of vaccines in March, 2020, to the first vaccine injections taking place by the end of December, 2020. To date, a second wave of new vaccine types includes 105 vaccines in clinical development and 184 in preclinical development.
Although the first set of 11 vaccines currently being used in vaccination roll-outs in several countries have proven to decrease infection and mortality rates, not all vaccines confer the same level of protection against current predominant variants such as B.1.1.7, B.1.351 and B.1.1.28/B.1.1.248 (Table 5 A-D).
While the safety of all approved vaccines in Canada continues to perform according to expected low levels of overall non-serious and serious event reports (see Table 5 A-B), there appear to be important distinctions. These comprise:
As has been the case in other countries, vaccination hesitancy across Canada during early vaccination roll-outs also decreased initial vaccination rates. On the basis of such hesitancy, provinces, such as Ontario, opted to endorse the mixing of two different vaccines. Individuals who had received a first dose of the Oxford-AstraZeneca DNA adenoviral vector vaccine received Pfizer-BioNTech or Moderna/NIH mRNA vaccines for their second vaccination dose. This vaccination strategy seems to have positively impacted vaccination rates, leading to a total of 71% of the population having received one dose (16%) or two (55%) as of July 24, 2021.
Due to the continuous emergence of new SARS-CoV-2 variants and close to 200 million people infected with SARS-CoV-2, it is highly likely that additional vaccination cycles will be required. Current desirable adjustments to the vaccines currently being used and developed should take into account an increased frequency of the NTD mutation L18F in both B.1.1.7 and B.1.351 together with the fact that NTD mutations seem to confer reduced sensitivity to neutralising antibodies[90]. Apart from addressing NTD mutations, the onset of potential undesirable side effects, in particular those related to thrombotic thrombocytopenia[97] and autoimmune related disorders[98,99], should also be taken into account.
As we may face the emergence of a SARS-CoV-2 super-variant, SARS-CoV-2 Variants of High Consequence and, possibly, a new Sarbecovirus lineage, the proven safety and efficacy of recent innovations in vaccinology by design using powerful technologies such as mRNA, chemical synthesis and peptide nano-engineering will assist us to lessen the impact of these threats to global health and the inevitable loss of a substantial number of human lives.
Acknowledgments
The authors wish to acknowledge the substantial and pioneering contributions of Dr Javier Pulido Alor, with the support of Enriqueta Cejudo Bretón, to the early detection, control of community transmission and treatment of tuberculosis.
For reprints, please use the contact form, below, or address the corresponding author, listed above.