
Journal of Entrepreneurial Health Sciences and Innovation (JEHSI)™
COVID-19: A Dynamic Multifactorial Model on the Origins of
SARS-CoV-2 S and L Types while Addressing the Potential Alternative
Vaccine-Derived Emergence of SARS-CoV-2 Progenitor
Dr Gabriel Pulido-Cejudo1,2*, Peter Humphries2, Dr Bratati Kar1,2, Dr Khadija El Abdaimi1,2 and Dr Abraham Pulido-Cejudo3
1International Centre for Advancement of Health Regional Innovation and Science, ICAHRIS
*corresponding author, gabriel.pulido-cejudo@icahris.org
2Canadian Federation of Breast Diseases, CFBD
3Hospital General de México
Abstract
Finding the origin of SARS-CoV-2, the putative infectious agent related to the ongoing COVID-19 pandemic outbreak, is crucial to mount a comprehensive and enduring response not solely for the current outbreak but for similar emerging virulent infections.
Based upon the most current and encompassing advances describing the potential origin of SARS-CoV-2, we recently advanced the use of lysosomotropic agents, gene editing and nNE adjuvant biogenic clones to hamper SARS-CoV-2 transmission and infectivity. The successful outcome of this clinical approach relies upon the known similarities found between SARS-CoV Spike Receptor Binding Domain (RBD) and that of SARS-CoV-2 as well as upon their common cellular entry to host cells through Angiotensin Converting Enzyme 2 (ACE2)-pH-dependent endocytosis.
Vis-à-vis the origin of SARS-CoV-2, the substantial level of homology shared between the SARS-CoV-2 and SARS-CoV RBDs and their common (ACE2)-pH-dependent endocytosis pathway are crucial because they appear to regulate both cross-species and human-to-human transmissions. Specifically, the amino acid sequence identity common between the SARS CoV-2 Spike peptide and that of SARS-CoV is close to 76.47%. Regardless of this functional and discrete high sequence homology, overall, SARS-CoV-2 and SARS-CoV share a lesser extent of homology (79.6%) when compared to that found between SARS-CoV-2 and bat SARSr-CoV RaTG13 (96.2%). This has led many to believe that bats are the original hosts; however, it is known that binding of SARSr-CoV RaTG13 Spike peptide to ACE2 is not as efficient as that of SARS CoV-2 Spike peptide. This calls for a closer examination of the most plausible origin of SARS-CoV-2 that might explain its higher binding capacity to ACE2 as well as its higher rates of transmission and pathogenicity by comparison to SARS-CoV and MERS-CoV.
Based upon the current lack of a definitive identification of an intermediate animal reservoir capable of conferring the adaptive changes required for efficient ACE2 binding, transmission and infectivity, we have previously proposed that a direct transmission of bat SARSr-CoV RaTG13 from bats to humans could have taken place, leading to the subsequent human adaptive changes of this ancestral precursor that were required for successful viral infectivity, replication and transmission. This series of events might have enabled the emergence of SARS-CoV-2 with its current level of global virulence.
In this article, we further assess this possible model, advancing the concept of a human-to-human superspreader-mediated transmission. We also address the most current and ongoing assessment of the possible emergence of SARS-CoV-2 through experimental processing of coronavirus-based vectors potentially used for the design of what could have been a universal vaccine against HIV, Malaria and, perhaps, other pathogens of current global public health intelligence priorities.
Keywords: Origin SARS-CoV-2 S and L Types, SARSr-CoV RaTG13, superspreader, COVID-19, zoonotic transmission, hibernaculum, intraspecies transmission, interspecies transmission, HIV, Malaria, Tuberculosis, Angiotensin Converting Enzyme 2 (ACE2), Receptor Binding Motif (RBM), S1-S2 subunits, Furin (S-Pro-Arg-Arg-Ala-R) Cleavage Site, Mutation Rates, Multifactorial Model, Gene Editing, Lysosomotropic Agents, nNE adjuvant biogenic clones, SARS-CoV-2/(ACE2)-pH-dependent endocytosis.
Article
Introduction
The fast pace of output of global scientific contributions has resulted in the rapid identification, sequencing and understanding of some of the most crucial cellular and intracellular infection pathways of SARS-CoV-2. These include the structural characterization of the SARS-CoV-2 Spike peptide Receptor Binding Domain (RBD) and of its strong binding to its putative Angiotensin Converting Enzyme 2 (ACE2) receptor through its Receptor Binding Motif (RBM), as well as its structural and functional resemblance to bat SARSr-CoV RaTG13. Some of the most relevant aspects of these SARS-CoV-2 characteristics have been previously described in depth, including the potential pH-dependent endocytic-mediated intracellular processing of SARS-CoV-2/ACE2 Receptor complexes leading to intracellular processing, sustained viral replication and transmission[1-6].
In reference to the most likely origin of SARS-CoV-2, together with its sustained airborne infectivity and transmission through aerosols and droplets, there are various known structural, functional and pathogenic properties of SARS-CoV-2 and of SARSr-CoV RaTG13 that should be taken into account in an effort to reconstruct the adaptive changes that one of the Severe Acute Respiratory Syndrome-Related Coronavirus (SARS-CoVs) ancestors should have undergone prior to the emergence of the highly contagious SARS-CoV-2 L Type. Some of these properties are summarized below and were used to articulate our Dynamic Multifactorial Model on the Origins of SARS-CoV-2.
Structural, Functional and Pathogenic Properties of SARS-CoV-2 and of SARSr-CoV RaTG13
There appears to be an overall high degree of genome sequence identity (96.2%) shared between bat SARSr-CoV RaTG13 and SARS-CoV-2; there are also patches of homology around their corresponding Receptor Binding Domains, which are both longer than any other SARSr-CoVs[1-6].
Full-length sequences isolated from five different patients at very early stages of the SARS-CoV-2/COVID-19 pandemic outbreak emerging from the city of Wuhan in central China were almost identical. Sharing 99.9% of sequence homology between each other, these strains originally identified as new coronavirus 2019-nCoV, were officially renamed as SARS-CoV-2[1,2,7-9].
Regardless of the high degree of genome sequence identity between SARSr-CoV RaTG13 and SARS-CoV-2, there are important functional differences at the protein level, particularly within their corresponding Receptor Binding Domains (RBDs). The RBDs from SARSr-CoV RaTG13 and SARS-CoV-2 show 85% of homology, sharing only one out of six of the amino acid residues needed for optimal binding to the putative Angiotensin Converting Enzyme 2 (ACE2) receptor favouring optimal binding by SARS-CoV-2[8].
SARS-CoV-2 and SARSr-CoV RaTG13 share a 93.1% nucleotide identity within their corresponding S gene, and both are longer than any other SARSr-CoVs[1,10]. Despite possessing three short insertions in the N-terminal domain and changes in four out of five of key amino acid residues in the Receptor Binding Motif (RBM) in comparison to SARS-CoV, the 3D structure and binding free energy between SARS-CoV-2 S-protein and human ACE2 remains high (-50.6 kcal/mol)[1,10]. This indicates that both have the innate ability to infect susceptible human cells through their binding capacity to ACE2 receptor[1,10]. In particular, the presence of Glutamine (Gln493) and Asparagine (Asn501) amino acid residues within the Receptor Binding Motif (RBM) appears to be crucial, not only for binding to ACE2 but also for enabling the acquired ability to facilitate human-to-human transmission[1,11].
Airborne and human-to-human transmission of SARS-CoV-2 through aerosols and droplets has been fairly well established; the virus is capable of gaining access to a variety of human cells other than lower respiratory tract hosts cells through binding and endocytic intracellular-pH-dependent processing of SARS-CoV-2/ACE2 receptor complexes[1,2,6-8]. Permissive cells of interest, in which there is the potential for infection, other than human pulmonary, kidney and intestinal cells, include neuronal and brainstem cells, which may explain the onset of neurological disorders such as loss of taste (ageusia), loss of sense of smell (anosmia) and confusion, present in some COVID-19 patients[12-14].
SARS-CoV-2 can gain entry into domesticated animals such as chickens, dogs, ducks and pigs. However, viral replication in these animals appears to be limited, with no proven infectivity in rats and mice. In contrast, successful viral replication seems to take place in cats and ferrets, eliciting infectivity[15]. In particular, cats are susceptible to airborne-induced infection, and transmission to naïve cats has been documented[15].
Although an intermediate reservoir host of SARS-CoV-2 has not being identified, bats are, beyond a doubt, the main reservoir of an array of Betacoronaviruses (β-CoVs) from which SARSr-CoV RaTG13, the presumed ancestor of SARS-CoV-2, could have emerged through adaptive evolution. Bats are also known to be the reservoir of several pathogenic coronaviruses such as SARS-CoV (Severe-Acute Respiratory Syndrome), MERS-CoV (Middle-East Respiratory Syndrome), PEDV (porcine epidemic diarrhea) and SADS-CoV (Swine acute diarrhea syndrome)[16]. Not being clinically affected by either natural or experimental infections, they are considered to be the ancestral hosts for several coronavirus (CoVs)[16].
Apart from wild animals sold for medicine and as bushmeat within the Huanan Seafood Wholesale Market in the city of Wuhan at the epicentre of the ongoing COVID-19 pandemic outbreak, there are numerous caves and caverns in the Province of Hubei lodging various bat colonies, some of which are in relatively close proximity to the capital city. A great number of comprehensive publications documenting the findings of what could represent thousands of coronavirus species have established that bat caves are at the epicentre of their ancestral evolution, bats being the natural reservoir of human coronavirus precursors such as SARSr-CoV RaTG13 from which SARS-CoV-2 is believed to have derived[17-19].
The addition of a four amino acid sequence (S-Pro-Arg-Arg-Ala-R) between the S1 and S2 Subunits of the SARS-CoV-2 Spike glycoprotein represents a unique Furin cleavage site motif which is not present in any coronavirus of the same clade[20,21]. Due to the ubiquitous presence of Furin in various human tissues and the potential need for a proteolytic-mediated immediate-early event of SARS-CoV-2 viral entry, it is possible that this sequence relates to its transmission capacity and infectivity.
Although no firm in vivo mutation rates for SARS-CoV-2 seem to be available, to date, based upon those reported for SARS-CoV and for other RNA viruses[22], the SARS-CoV-2 mutation rate could be expected to be ≥ 6.0 x 10-6 nucleotide substitutions per site daily or 0.2 mutations per genome per day. Based upon our previous report, indicating that the first known case of COVID-19 might have taken place in or around December 8[2], the relative amount of mutations could have already reached 30 mutations per genome in asymmetrical proportion, due to the fairly rapid human intervention.
It is likely that, as a result of the previously described property regarding the in vivo asymmetrical mutations of the ancestral SARS-CoV-2, the recently described SARS-CoV-2 S Type and the SARS-CoV-2 L Type have emerged[23]. As a result of such an asymmetrical mutation pathway, SARS-CoV-2 L Type has 70% prevalence whereas SARS-CoV-2 S Type accounts for 30% of prevalence. In addition, SARS-CoV-2 L Type appears to be more aggressive, possessing a faster spreading pace than that found in SARS-CoV-2 S Type[23]. Interestingly, SARS-CoV-2 S Type is evolutionary older, closer to the ancestral progenitor and possessing a less aggressive phenotype[23].
Since the emergence of SARS in 2002/2003, extensive biological sampling and characterization of bat fluids in search of potentially pathogenic bat coronaviruses has been undertaken by various research groups[24,25]. Contrary to the use of heavily attenuated murine coronavirus genomes to construct biosafe coronavirus-based vaccine vectors simultaneously expressing stimulatory cytokines to target dendritic cell response together with epitopes of interest such as Melan-A as well as lymphocytic choriomeningitis virus glycoprotein[26], to date, no heavily attenuated bat coronavirus genomes have been reported. It is conceivable that a biosafe bat coronavirus-based vaccine vector could have been designed containing multiple epitopes of interest for vaccine development against some of the most relevant pathogens of interest, such as Plasmodium malariae, falciparum, vivax, ovale, Micobacterium tuberculosis and HIV, which, together, account for more than 5 million deaths per year[27]. However, evidence that such a system has been engineered has not been reported.
COVID-19: A Dynamic Multifactorial Model of the Origins of SARS-CoV-2 S and L Types
Supported by the evidence and careful review of information available during the early phases of the current SARS-CoV-2/COVID-19 pandemic outbreak, together with the lack of a consistent identification of pangolins as an intermediate reservoir of human SARS-CoV-2[28,29], in a previous report, we had anticipated that a potential intermediate animal reservoir of SARS-CoV-2 could have been present within the Huanan Seafood Wholesale Market located in the city of Wuhan[2]. It was reported that, in addition to seafood, wild animals were also available within this market which equally happened to be the workplace of approximately 27 vendors who had been hospitalised with what is now known as COVID-19[2].
Although there is the prospect of a future finding of an intermediate animal reservoir of SARS-CoV-2, in this article, we further advance the concept of a dynamic multifactorial model of the origin of SARS-CoV-2 based upon some of its structural, functional and pathogenic characteristics and resemblances between SARS-CoV-2 and its ancestral progenitor, SARSr-CoV RaTG13.
At the centre of this model, it is highly likely that SARS-CoV-2 emerged from a process of adaptive evolution of its bat SARSr-CoV RaTG13 Severe Acute Respiratory Syndrome-Related Coronavirus (SARS-CoV) ancestor. Indeed, bats have been the reservoirs of several pathogenic coronaviruses such as SARS-CoV (Severe-Acute Respiratory Syndrome), MERS-CoV (Middle-East Respiratory Syndrome), PEDV (porcine epidemic diarrhea) and SADS-CoV (Swine acute diarrhea syndrome). Unaffected, clinically, by neither natural nor experimental infections, they are considered to be the ancestral hosts for several coronaviruses (CoVs), including SARSr-CoV RaTG13[1,2,16]. As illustrated in Figure 1, our proposed dynamic multifactorial model of the origin of SARS-CoV-2 encompasses four major components:
- The central role of bats in the origins of SARS-CoV-2, not solely due to its high level of homology with bat SARSr-CoV RaTG13 but, also, due to the proximity of bat populations living in caverns and caves within the Hubei province close to the capital city of Wuhan at the epicentre of the ongoing COVID-19 pandemic outbreak. It is possible that exposure and coronavirus spillover from bat to humans might have taken place due to the disturbance of bat ecological niches, driven by regional anthropogenic activities, bat trading and consumption[2,16,30].
- Intraspecies and interspecies transmission, replication and adaptive evolution of bat CoVs, in particular of SARSr-CoV RaTG13 / SARS-CoV-2 S Type, depending on the ubiquitous expression of Angiotensin Converting Enzyme 2 (ACE2) / ACE2-like receptors of potential animal host systems and environmental and related selective pressures.
- Airborne infectivity and human-to-human transmission through aerosols and droplets, potentially accelerated by the presence of human superspreaders[31] and, perhaps, related to infected asymptomatic wild animal traders in the vicinity of Huanan Seafood Wholesale Market. SARSr-CoV RaTG13 spillover into humans might have taken place prior to December 8, 2019, when the first case of SARS-CoV-2 was apparently reported[2].
- Inadvertent coronavirus (SARSr-CoV RaTG13) spillover from bats to humans during field sampling and transportation of bat biological fluids and during laboratory expansion of clones of interest through selective viral cultures in permissive cells such as African green monkey Vero kidney cells[24, 25]. Although a laboratory spillover of an ancestral progenitor of SARSr-CoV RaTG13 / SARS-CoV-2 S Type could have taken place, it is highly unlikely that cell culture and viral expansion of such an ancestral progenitor alone could have resulted in the emergence of SARS-CoV-2. In vitro cell culture mutation rates of SARS-CoV, the closest coronavirus to SARS-CoV-2, have been reported to be negligible[22].
Gene editing, for the purpose of engineering a biosafe bat coronavirus-based vaccine vector against Plasmodium malariae, falciparum, vivax, ovale, Micobacterium tuberculosis and HIV, leading to human spillover of SARS-CoV-2 ancestor remains a path yet to be elucidated and documented.
Although an intermediate reservoir host of SARS-CoV-2 has not being identified, bats are, demonstrably, the main reservoir of an array of Betacoronaviruses (β-CoVs) from which SARSr-CoV RaTG13 could have emerged through adaptive evolution. Such a process could have taken place through an intermediate animal reservoir after bat zoonotic transfer, or, as we have previously suggested, adaptive evolution could have taken place by means of human-to-human transmission after bat zoonotic transfer of SARSr-CoV RaTG13 ancestral progenitor of SARS-CoV-2 S Type.
Direct zoonotic transmission from bat to humans takes into account the ability of SARSr-CoV RaTG13 Binding Motif (RBM) to bind to ACE2 receptors based upon its potentially similar 3D structure and binding free energy capacity to that reported between SARS-CoV-2 S-protein and its human ACE2 receptor[1,10]. This would imply that they both have the innate capacity to infect susceptible human cells through their binding capacity to ACE2 receptors[1,10].
In addition, based upon the in vivo mutation rates reported for SARS-CoV[22] and, hence, the assumption of a mutation rate for SARS-CoV-2 of at least 0.2 mutations per genome per day, the bat SARSr-CoV RaTG13 ancestral progenitor could have the capacity to transition into SARS-CoV-2 S Type at a viable rate after bat zoonotic transmission into humans, advancing from a precarious infectious agent to a more aggressive pathogen.
This possibility is further supported by the isolation and characterisation of SARS-CoV-2 S and L Types[23]. Based upon these findings, it appears that SARS-CoV-2 S-Type, which was found to be closer to the ancestral progenitor, is less prevalent (30%) and less aggressive than the L Type, which has a prevalence of 70%, demonstrating the capacity to spread faster and possessing a sequence further apart from the ancestral sequence. Interestingly, it has also been found that the higher prevalence of the SARS-CoV-2 L Type in comparison to the SARS-CoV-2 S Type decreased with time, after the initial outbreak, explained by fairly rapid human intervention[23].
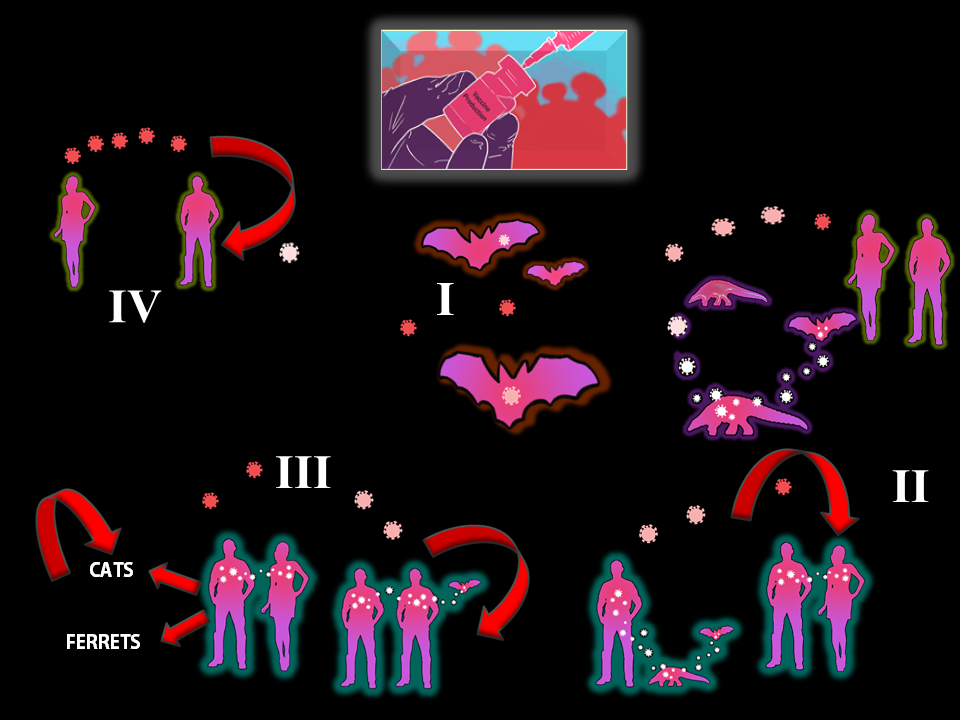
Figure 1 A Dynamic Multifactorial Model on the Origin of SARS-CoV-2 S and L Types
As illustrated in this figure, there are at least four critical components (I-IV) that should be taken into consideration whilst addressing the origins of SARS-CoV-2 S and L types and, perhaps, of other emerging infectious SARSr-CoVs. Although each individual component (I-IV) has already been described, this figure illustrates (red arrows) the dynamics of potential interspecies (humans to cats / ferrets) and intraspecies (cats-to-cats / ferrets-to-ferrets) transmission and the adaptive evolution of the SARS-CoV-2 ancestral progenitor, the bat SARSr-CoV RaTG13. The adaptive evolution of the ancestral progenitor, bat SARSr-CoV RaTG13 (white) into the current SARS-CoV-2 S Type (pink) and SARS-CoV-2 L Type (red) could have occurred through direct zoonotic transmission from bat to humans and subsequent adaptive intraspecies transmission through human superspreaders[31], perhaps related to infected asymptomatic wild animal traders in the vicinity of Huanan Seafood Wholesale Market. Gene editing for the purpose of engineering a biosafe bat coronavirus-based vaccine vector against Plasmodium malariae, falciparum, vivax, ovale, Micobacterium tuberculosis and HIV leading to human spillover of SARS-CoV-2 ancestor remains a path yet to be elucidated and documented (IV). No reports of an inadvertent coronavirus (SARSr-CoV RaTG13) spillover through bat sampling, transportation and laboratory amplification have been published.
The finding of SARS-CoV-2 S and L Types related to the initial differences in prevalence and level of aggressive phenotypes together with the reported changes in their subsequent prevalence after the SARS-CoV-2 outbreak, potentially due to fairly rapid human intervention[23], demonstrates the multifactorial nature of selective pressures affecting the origin and continuous adaptive evolution of SARS-CoV-2.
A greater level of refinement and support for our model regarding a direct zoonotic transmission of bat SARSr-CoV RaTG13 from bats to humans could be gained through sequence analysis of archive samples obtained from affected individuals in Hong Kong, Japan and Thailand during initial stages of the ongoing SARS-CoV-2 pandemic outbreak where exported cases of SARS-CoV-2 were reported[7].
Genomic analysis of such samples within the Receptor Binding Motifs and, in particular, close to the unique (S-Pro-Arg-Arg-Ala-R) Furin cleavage site motif, previously described, could shed light onto progressive adaptive evolutionary changes between SARS-CoV-2 S and L Types and the possible existence of ancestral sequences intermediate between bat SARSr-CoV RaTG13 and SARS-CoV-2 S Type. Such findings could lead to the discovery of a missing link between SARSr-CoV RaTG13 and SARS-CoV-2 S Type, further supporting the emergence of SARS-CoV-2 by means of a direct zoonotic transmission of bat SARSr-CoV RaTG13 to humans. A direct zoonotic transmission of SARSr-CoV RaTG13 to humans, potentially leading to the emergence of SARS-CoV-2 S Type, is consistent with the unique, longer S genes sequences found in both SARSr-CoV RaTG13 and SARS-CoV-2 when compared to those found in currently known SARSr-CoVs[1].
As we previously reported[2], Wuhan authorities closed the Huanan Seafood Wholesale Market where a variety of exotic wild animals were apparently sold for human consumption, inspecting and ordering the immediate sanitization and closure of the market. It would be of interest to determine if an exact inventory of wild animals found was performed and, most importantly, if swab samples of the animals and of public surfaces of the market took place. If such a detailed examination was conducted, it would be of interest to make public any sequences found in the animals and the public surroundings. In order to trace back the origins of SARS-CoV-2 (S and L Types), it would be ideal to examine samples from known animal traders and from all possible merchants and their costumers between early December, 2019, and April, 2020.
Anthropogenic Activities Influencing the Emergence of Pathogenic Diseases: The Protective Fast Replicating and Intraspecies Transmissible Bat Viral Biome
The current consensus indicates that the ancestral origin of SARS-COV-2 could, potentially, be traced back to the bat SARSr-CoV RaTG13 Betacoronavirus 2 β lineage[1,2], bats being the ancestral or original hosts. From a broader perspective, it has been considered that wildlife animal species account for an estimated 75% of infectious diseases affecting humans, of which 58% are zoonotic[32,33].
Bats, in particular, fruit bats, are the natural hosts of a considerable number of emerging viruses. Estimated in the thousands, some viruses naturally hosted in bats have resulted in life-threatening disease outbreaks affecting humans. These involve a string of viruses such as Ebola, Nipah, Marburg and several coronaviruses including SARS, MERS and SARS-CoV-2[1,2,16,32-35]. Table 1 summarizes some of the most relevant types of viruses and the species of bats found to be their natural hosts.
It is believed that bats have developed a complex and efficient mechanism to suppress the infectivity of a great number of viruses, becoming their natural ancestral hosts, perhaps, for thousands or millions of years after their emergence approximately 55 million years ago with the appearance of Hassianycteris kumari[37-39].
| Virus | Family | Genus | Bat species (common name) |
| Ebola | Filoviridae | Ebolavirus | Miniopterus inflatus (greater long-fingered bat) |
| Marburg | Filoviridae | Marburgvirus | Rousettus aegypti (fruit bats of the Pteropodidae family) |
| Nipah | Paramyxoviridae | Henipavirus | Pteropus hypomelanus (variable flying fox), Pteropus vampyrus (large flying fox), Pteropus lylei (Lyle's flying fox) |
| Hendra | Paramyxoviridae | Henipavirus | Pteropus alecto (black flying fox), Pteropus poliocephalus (gray-headed flying fox), Pteropus scapulatus (little red flying fox), Pteropus conspicillatus (spectacled flying fox) |
| SARS-CoV | Coronaviridae | Betacoronavirus | Rhinolophus sinicus (Chinese horseshoe bat), Rhinolophus pearsonii (Pearson's horseshoe bat), Rhinolophus macrotis (big-eared horseshoe bat), Rhinolophus ferrumequinum (greater horseshoe bat) |
| MERS-CoV | Coronaviridae | Betacoronavirus | Rhinolophus sinicus (Chinese horseshoe bat), Rhinolophus pearsonii (Pearson's horseshoe bat), Rhinolophus macrotis (big-eared horseshoe bat), Rhinolophus ferrumequinum (greater horseshoe bat) |
| SARS-CoV-2 | Coronaviridae | Betacoronavirus | Rhinolophus sinicus (Chinese horseshoe bat), Rhinolophus pearsonii (Pearson's horseshoe bat), Rhinolophus macrotis (big-eared horseshoe bat), Rhinolophus ferrumequinum (greater horseshoe bat) |
Table 1 Bats are the Natural Hosts of Several Virus Types Severely Affecting Human Health
Bats are considered the ancestral hosts of numerous and diverse viruses affecting human health[16,30,32-35]. Although it is believed that bats have developed the capacity to effectively suppress the infectivity of a great diversity of viruses, including those in Table 1, being their natural hosts, perhaps, for thousands of years, recent highly disruptive human activities affecting their natural environments, including disturbances of their winter hibernoculum[36] have led to a greater level of direct exposure of humans to their saliva, urine and feces carrying highly virulent strains. Some of them have caused recent severe disease outbreaks, including the ongoing SARS-CoV-2 pandemic outbreak. It is also conceivable that recent, disruptive, anthropogenic activities have also affected the ability of bats, themselves, to neutralize the infectivity of some non-viral infectious agents transmitted from humans to bats. Regarding viral pathogens, the evolutionary capacity of bats to cope with multiple viral infections appears to be related to an attenuated synthesis of Interferons through a decreased response mediated by the Stimulator of Interferon Genes (STING) / Transmembrane Protein 173 (TMEM173) peptide[40] as well as to the complete abrogation of the PYRIN and HIN domain (PYHIN) gene family[41]. The PYHIN gene family has been associated with the expression of immunosensors capable of activating inflammasome and / or interferon-mediated inflammatory cascade cellular processes. They consist of immune sensors of intracellular DNA that have been related to the activation of inflammasome and / or interferon-mediated inflammation[42].
From a sheer evolutionary standpoint, the exquisite balance achieved by bats through millions of years of adaptive evolution has converted otherwise human pathogenic viruses into part of what could be considered to be their protective, bat-replicating, intraspecies-transmissible biome, shielding against predators, including humans.
Nonetheless, years of anthropogenic erosion of bat ecosystems, including their winter hibernaculum[36], destruction of their natural habitats, wide use of their feces as fertilizers (bat guano), their illegal trading for believed medicinal remedies and bushmeat[2,30] and constant, disruptive tourism-related activities, biological sampling and testing have ignited a global health imbalance affecting humans, domestic animals and bats alike.
Combined, these multifactorial components influencing the adaptive evolution of viruses hosted within bats can lead to the origin of several emerging Betacoronaviruses including SARS-CoV-2 S Type. In the absence of the clear identification of an as yet elusive intermediate host between bats and humans, our model of a direct zoonotic transmission from bats to humans takes into account the close functional and structural homology between bat SARSr-CoV RaTG13 and SARS-CoV-2 S Type, as described in previous sections. It is also based upon the aforementioned disruptive anthropogenic activities that have significantly increased human exposure, not only to bats but to an array of wild animals capable of the direct zoonotic transmission of several pathogens affecting human health[32-37,43]. Apart from affecting human health, these disruptive anthropogenic activities have already put at risk 167 different bat species, particularly due to opportunistic, year round hunting practices[30,44-46].
Indiscriminate hunting and trading for bushmeat has also critically endangered at least 72 terrestrial mammals, with the most affected species including pangolins, platypus and echidnas, odd-toed ungulates, primates and even-toed ungulates[45]. It is most likely due to a high level of hunting, trading and consumption of some of these mammals in China that pangolins were initially considered to function as the potential intermediate hosts of SARS-CoV-2[28].
To date, the possible role of pangolins as intermediate hosts for SARS-CoV-2 remains improbable due to the absence of the (S-Pro-Arg-Arg-Ala-R) Furin cleavage Motif found in SARS-CoV-2 but not within some of the pangolin sequences analyzed[29]. As previously mentioned, this unique sequence might be related to SARS-CoV-2 immediate viral entry processing, modulating its transmission capacity and infectivity[20,21].
In relation to human laboratory-based monitoring activities of pathogens harboured within various bat populations, bat viruses have been isolated from numerous bat species, worldwide, and, most intensively, since the beginning of the first SARS CoV Betacoronavirus outbreak in 2002 / 2003. One of the most recent and encompassing database (DVatVir) describes more than 4100 bat-linked animal viruses included within 23 viral families isolated from 196 bat species within 69 countries[47].
This rather comprehensive investigation of bat-linked viruses is, undoubtedly, accompanied by human exposure while conducting controlled activities using live samples from bat tissues and body fluids such as handling, processing, using, storing, producing, accessing, transferring, exporting / importing, releasing and disposing of such samples. Hence, there is, naturally, the likelihood of human exposure to and laboratory acquire infections (LAIs) from an array of bat-viruses, including Betacoronaviruses.
Although currently, in Canada, SARS-CoV-2 has been classified as a Risk Group 3 (RG3) human pathogen by the Centre for Biosecurity, the current level of risk to humans of the more than 4,100 bat-associated animal viruses remains to be determined and, perhaps, in some cases, reclassified. Apart from securing the appropriate licensing status of national and international laboratory facilities handling, working, culturing and cryopreserving animal and bat viruses, such as those included within the (DVatVir) Database by their corresponding health authorities, it is of relevance to make sure that adequate laboratory Containment Levels (CL) are also used.
Currently, CL2 facilities are required for viral concentration, inactivation, extraction, molecular testing and performance of antigen / antibody immunoassays for SARS-CoV-2, whilst CL3 facilities are required for culturing, propagation, processing of cultures and preparatory activities prior to in vivo activities (Centre for Biosecurity). Although these comprehensive levels of biosecurity practices reduce the chances of laboratory spillover of animal viruses, including those harboured and isolated from bats, into humans, it would be of interest to secure sentinel monitoring testing and levels of exposure of individuals involved in the amplification, cell propagation and isolation of animal viral cultures.
With respect to gene editing laboratory activities for the purpose of engineering a biosafe bat coronavirus-based vaccine vector against Plasmodium malariae, falciparum, vivax, ovale, Micobacterium tuberculosis and HIV leading to human spillover of the SARS-CoV-2 ancestor, that remains a path yet to be elucidated and documented. No reports of an inadvertent coronavirus (SARSr-CoV RaTG13) spillover through bat sampling, transportation and laboratory amplification or gene editing has been published.
Conclusions
Almost two months ago, in our previous publication concerning the potential inhibition of community transmission of SARS-CoV-2 / COVID-19 with the combined use of lysosomotropic agents, gene-editing and the adjuvant use of biogenic clones, the incidence of COVID-19 had attained 198,348 cases, leading to 7,979 deaths[2]. To date, 4,381,814 people have been infected, causing the death of 294,758 ill patients.
Thus, finding the origin of SARS-CoV-2, the causative infectious agent of the sustained COVID-19 pandemic outbreak, is central to halt its rapid spread as well as to mount an encompassing and enduring response, not just against the current outbreak but for emerging Betacoronavirus virulent infections. On the basis of both the structural and functional characteristics shared between bat SARSr-CoV RaTG13, human SARS-CoV-2 and, most likely, SARS-CoV-2 S Type, a dynamic multifactorial model of the origins of SARS-CoV-2 S and L Types is provided.
Of the two most commonly found viral zoonotic modes of transmission from the harbouring, original hosts to humans, transmission through an intermediate animal host and direct zoonotic transmission from ancestral hosts to humans, we proposed that the most likely origin of SARS-CoV-2 is the direct zoonotic transmission of the ancestral SARSr-CoV RaTG13 from bats to humans. This conclusion is based upon the similar structural characteristics shared only between SARSr-CoV RaTG13 and SARS-CoV-2 S, such as:
- A high degree of genome sequence identity (96.2%) shared between bat SARSr-CoV RaTG13 and SARS-CoV-2, together with the existence of patches of homology around their corresponding Receptor Binding Domains;
- The presence of 85% of homology within their Receptor Binding Domains (RBs), which are longer than any other SARSr-CoVs;
- The presence of one out of six amino acid residues needed for optimal binding to their putative Angiotensin Converting Enzyme 2 (ACE2) receptor, favouring optimal binding by SARS-CoV-2 in comparison to SARSr-CoV RaTG13;
- The presence of unique (S-Pro-Arg-Arg-Ala-R) Furin cleavage sites within the Receptor Binding Motifs absent in any other SARSr-CoVs, including those isolated from pangolins which were initially proposed as the intermediate reservoir animal of SARS-CoV-2.
A dynamic transmission model from bats to humans also takes into account several anthropogenic disruptive activities that might have favoured an accelerated direct zoonotic transmission of ancestral bat SARSr-CoV RaTG13 from bats to a potential human superspreader. These include:
- Years of anthropogenic erosion of bat ecosystems, including their winter hibernaculum;
- Annihilation of their natural habitats, year round;
- Wide use of their feces as fertilizers (bat guano), driving the erosion of their habitats whilst exposing humans to an array of Betacoronaviruses, amongst several other type of viruses;
- The illegal trading of bats for believed medicinal remedies and bushmeat, as well as constant, disruptive, tourism-related activities, biological sampling and testing.
The compounded effect of these anthropogenic activities, together with the greater exposure of humans to bat body fluids and tissues and the remarkable bat intraspecies transmission and evolution of Betacoronaviruses that, like SARS-CoV-2, could potentially attain mutation rates ≥ 6.0 x 10-6 nucleotide substitutions per site, daily, would have been enough to enable human-to-human transmission and adaptive evolution of the ancestral bat SARSr-CoV RaTG13 into SARS-CoV-2 S Type, followed by human intraspecies adaptive changes of SARS-CoV-2 S Type into the more aggressive and highly transmissible SARS-CoV-2 L Type.
Our dynamic multifactorial direct zoonotic transmission model involving the interspecies spillover of bat SARSr-CoV RaTG13 ancestor from bats to humans without an intermediate animal host, leading to the origin of SARS-CoV-2, also envisages intraspecies adaptive evolution that not only accelerates human-to-human transmission but interspecies transmission from humans to domesticated animals, explaining the current findings of potential transmission, effective replication and infectivity of SARS-CoV-2 into cats and ferrets.
In closing, moving from a model of the potential origin of SARS-CoV-2 to the foundation of a future global sentinel programme to lessen the likelihood of emerging Betacoronavirus pandemic outbreaks and decrease the current impact of SARS-CoV-2 on the health of humans and, possibly, on that of some domesticated animals, such as cats and ferrets, prior to the successful deployment of safe and effective vaccines, it would be of interest to:
- Continue monitoring intraspecies SARS-CoV-2 transmission, including human-to-human transmission of S, L and emerging regional Types / Subtypes of SARS-CoV-2;
- Confirm interspecies human-to-domesticated animal SARS-CoV-2 transmission, replication and intraspecies infectivity between domesticated animals, in particular involving cats and ferrets;
- Articulate a global and concerted effort aimed at halting the further erosion and fragmentation of bat habitats and surrounding ecosystems, particularly in Africa, China, Oceania, Central America and South America, amongst other regions and countries known to trade in and perform predatory activities against a variety of bat species;
- Structure an international, permanent ban on global, regional and local wild animal trading for medicinal purposes, bushmeat and for any other activities affecting the survival of wild animal species, their habitats and ecosystems;
- Proceed with global, regional and local educational campaigns explaining the generational consequences of continuing to erode wild plant and animal ecosystems for untenable food, housing, wild animal trading, trophy hunting and other related anthropogenic activities that exacerbate human exposure to and transmission of zoonotic infectious diseases;
- Establish a concerted, international effort to protect bat cavern and cave habitats and hibernaculum throughout the calendar year, limiting access and biological sampling of bat tissues and body fluids. Instead, the assessment of potential human exposure to bat Betacoronaviruses and to other viral pathogens harboured by bats could be achieved by monitoring human populations living in close proximity to caverns and caves that shelter important populations of bats. Such sentinel screening could be effected based upon current, robust databases.
To conclude, it is hoped that the current SARS-CoV-2 pandemic outbreak has clearly exposed the severe consequences that could arise through an accidental spillover of gene-edited coronaviruses, in particular Betacoronaviruses. In their natural, non-manipulated forms, Betacoronaviruses have already been related to three significant infectious disease outbreaks -- SARS, MERS and, currently, the COVID-19 pandemic occurrence.
Acknowledgments
We thank Andrés Olmos Muraira and Mónica Muraira for the original illustration.
For reprints, please use the contact form, below, or address the corresponding author, listed above.